
A rare disease is defined by the European Union as a condition that affects fewer than 1 in 2,000 people within the general population. There could be as many as 7,000 rare diseases. Despite having a low incidence, the total number of Americans living with a rare disease is estimated at between 25-30 million. These figures highlight that while individual diseases may be rare, the total number of people with a rare disease is large, a fact that is often misunderstood by those unfamiliar with the rare disease community. [1]
Cyagen assists in the research of rare diseases and provides various mouse/rat models, including the Cep290 knockout mouse. This article focuses on the gene CEP290 and its related rare diseases, aiming to help interested researchers continue to build on scientific literature and knowledge.
Overview of CEP290 Gene
CEP290 (Centrosomal Protein 290) is a Protein Coding gene, encodes a centrosomal protein involved in ciliary assembly and ciliary trafficking. [2] This gene encodes a protein with 13 putative coiled-coil domains, a region with homology to SMC chromosome segregation ATPases, six KID motifs, three tropomyosin homology domains and an ATP/GTP binding site motif A. The protein is localized to the centrosome and cilia and has sites for N-glycosylation, tyrosine sulfation, phosphorylation, N-myristoylation, and amidation. Mutations in this gene have been associated with Joubert syndrome and nephronophthisis. The presence of antibodies against this protein is associated with several forms of cancer.
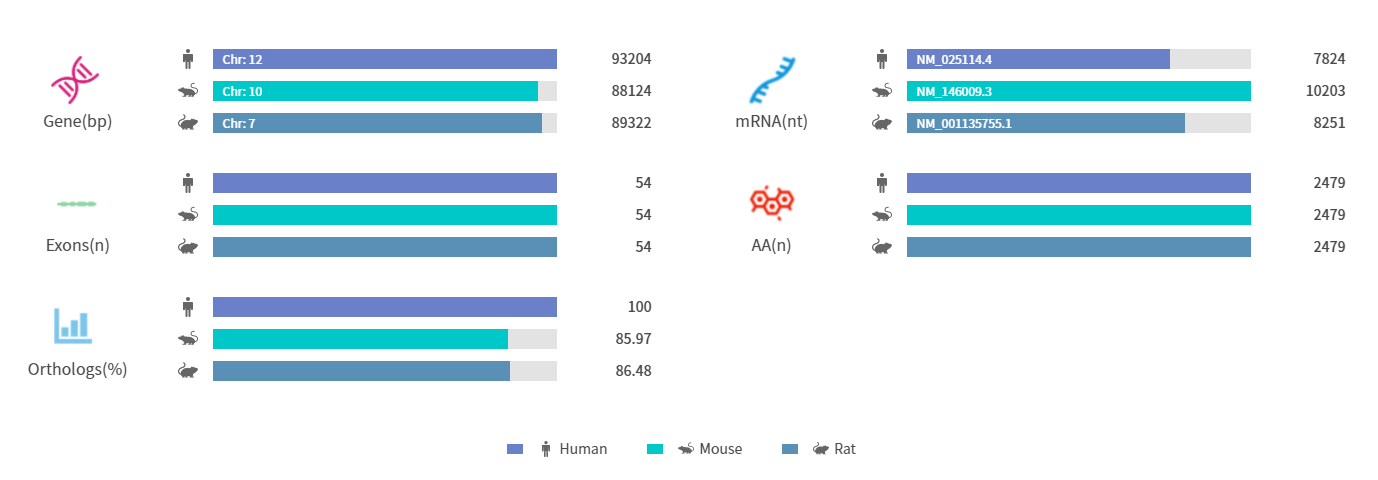
Figure 1. Gene information and sequence homology alignment of CEP290 [3]
(Source: Rare Diseases Data Center (RDDC))
Rare Diseases Associated with the CEP290 Gene
Leber congenital amaurosis (LCA) is the most common cause of inherited childhood blindness with roughly 3 out of 100,000 children around the world affected by the disorder. [4]
Symptoms of LCA appear within the first years of life, often resulting in significant vision loss and potential blindness.
Symptoms include:
▪ Significant vision loss
▪ Rapid involuntary eye movement
▪ No measurable electroretinogram activity due to loss of photoreceptor cells (the cells in the eye that take in light and help us see)
The most common form of the disease, LCA10, accounts for approximately 20-30 percent of all LCA patients. LCA10 is caused by mutations in the CEP290 gene; there are currently no treatment options available for LCA10.
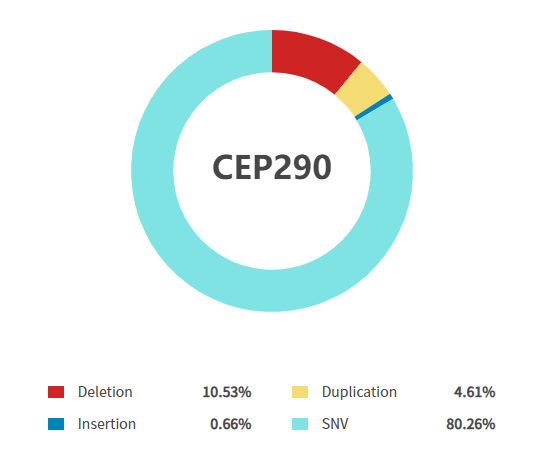
Figure 2: CEP290 gene mutation types resulting in onset of LCA10 [5]
(Source: Rare Diseases Data Center (RDDC))
In 2019, Maeder and colleagues have developed EDIT-101, an experimental genome-editing medicine designed to remove the abnormal splice donor site caused by the IVS26 mutation in CEP290 (which most commonly causes LCA10), and thereby restore normal CEP290 expression. In their study, the researchers showed that subretinal delivery of EDIT-101 in a human CEP290 IVS26 knock-in mouse model resulted in fast and sustained CEP290 gene editing. They also demonstrated productive gene editing in a comparable surrogate nonhuman primate vector at levels meeting the target therapeutic threshold. [6]
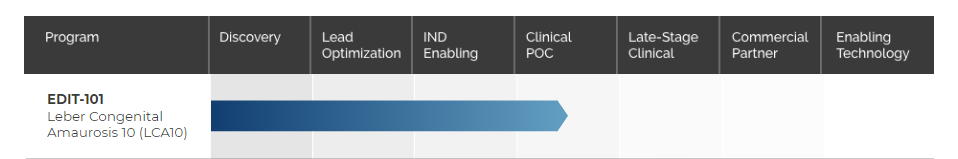
Figure 3. Expansive Pipeline for Gene Edited Medicines-EDIT-101 (Source: Editas Medicine)
Preliminary EDIT-101 clinical results demonstrated a favorable safety profile and encouraging signals of clinical benefit. Editas remains on track to complete dosing of the pediatric mid-dose cohort in the first half of 2022 and expects to initiate dosing of the pediatric high-dose cohort this year.
In any case, gene therapy brings hope for rare genetic diseases, let's wait and see how EDIT-101 performs next.
Joubert syndrome is a neurodevelopmental disorder, characterized by malformation of the mid and hindbrain leading to the pathognomonic molar tooth appearance of the brainstem and cerebellum on axial MRI.
Core clinical manifestations include hypotonia, tachypnea/apnea, ataxia, ocular motor apraxia, and developmental delay of varying degrees. In addition, a subset of patients has retinal dystrophy, chorioretinal colobomas, hepatorenal fibrocystic disease, and polydactyly.
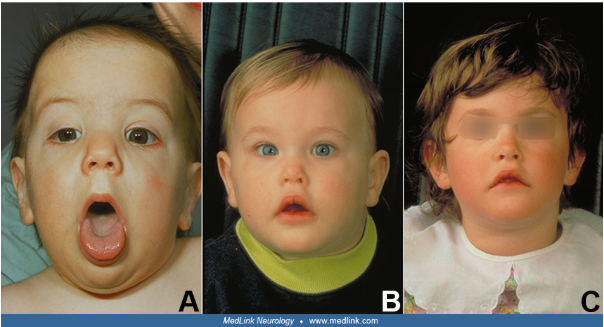
Figure 4. Clinical features in Joubert syndrome (Source: Medlink)
Currently, precision treatments for Joubert syndrome are only available as part of research studies (investigational). Gene therapy has been used to improve visual function for specific types of retinal dystrophy by direct retinal injection of expression vectors in humans. Future trials are likely to involve the CEP290 gene, a major cause of Leber congenital amaurosis and Joubert syndrome. Alternatively, antisense oligonucleotides can augment gene function by blocking aberrant splicing or by causing exons with deleterious variants to be skipped. These treatments are still in early development for Joubert syndrome but have been used for other disorders in humans such as spinal muscular atrophy (SMA). Future medications targeting specific pathways that are disrupted in Joubert syndrome may be beneficial, but this work is also in its infancy.
What Can Cyagen Do?
Over the past year, Cyagen has focused on developing educational resources and initiatives to assist the rare disease research community. To help inform our R&D of precise animal models for rare disease studies, we initiated a Rare Disease Model Collaboration Program in the first quarter of 2021. With this program, we aim to build a community of rare disease researchers with ideas for the next generation of animal models needed to advance their field of study.
References:
[1] FAQs About Rare Diseases https://rarediseases.info.nih.gov/diseases/pages/31/faqs-about-rare-diseases
[2] Coppieters F, Lefever S, Leroy BP, De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum Mutat. 2010 Oct;31(10):1097-108. doi: 10.1002/humu.21337. PMID: 20690115.
[3] Gene information and sequence homology alignment of CEP290 https://rddc.tsinghua-gd.org/details/gene?gene=2VRlB2
[4] den Hollander AI, Koenekoop RK, Yzer S, Lopez I, Arends ML, Voesenek KE, Zonneveld MN, Strom TM, Meitinger T, Brunner HG, Hoyng CB, van den Born LI, Rohrschneider K, Cremers FP. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006 Sep;79(3):556-61. doi: 10.1086/507318. Epub 2006 Jul 11. PMID: 16909394; PMCID: PMC1559533.
[5] CEP290 gene mutation types resulting in onset of LCA10 https://rddc.tsinghua-gd.org/details/disease?disease=x0kljG
[6] Maeder ML, Stefanidakis M, Wilson CJ, Baral R, Barrera LA, Bounoutas GS, Bumcrot D, Chao H, Ciulla DM, DaSilva JA, Dass A, Dhanapal V, Fennell TJ, Friedland AE, Giannoukos G, Gloskowski SW, Glucksmann A, Gotta GM, Jayaram H, Haskett SJ, Hopkins B, Horng JE, Joshi S, Marco E, Mepani R, Reyon D, Ta T, Tabbaa DG, Samuelsson SJ, Shen S, Skor MN, Stetkiewicz P, Wang T, Yudkoff C, Myer VE, Albright CF, Jiang H. Development of a gene-editing approach to restore vision loss in Leber congenital amau
We will respond to you in 1-2 business days.