
After more than 30 years of cancer research, many targeted therapies have been approved to treat a variety of tumors, and many more are under development or in early-stage clinical trials. However, the recurrence and drug resistance of different cancers makes drug development more difficult. The development of high-efficiency drugs against these two characteristics, cancer recurrence and drug resistance, has gradually become a hot research area for cancer therapies.
Mutations in oncogenes or tumor suppressor genes of single cells are the basis of tumorigenesis, a process in which a cell it loses its original cell identity. The complex tumor environment can be viewed as a dynamic, interacting system (tumor microenvironment) composed of single cells. Single-cell sequencing, capable of characterizing the genome of a single cell, has had a profound impact on the advancement of tumor biology and genetics.
What is Single-Cell Sequencing?
Single-cell sequencing refers to sequencing the genome or transcriptome of a single cell to obtain genomic, transcriptome, or multi-omics data information. The single-cell sequencing technique thereby reveals cell population differences and cell evolutionary relationships.
Single-Cell Sequencing VS Traditional Methods
Traditional sequencing methods are used in sequencing the genome or transcriptome of multiple cells, so the information on cellular heterogeneity cannot be analyzed. In contrast, single-cell sequencing can be used to detect heterogeneity between individual cells, distinguish small numbers of cells, and delineate cell maps.
We can compare single-cell sequencing and traditional sequencing through the following analysis. As shown in the figures below, among the 3 groups of cells, there was a significant change in the phenotype of one cell in Group 1, half of the cells in Group 2 had a certain degree of change, and all cells in Group 3 had weak phenotype changes. Using Western blotting (WB) to detect these three groups of cells, the bands may be consistent, and traditional sequencing methods may also fail to obtain significant differences between groups of cells.
Group 1
Group 2
Group 3

Western blot bands of three groups of cells
Sequencing results of three groups of cells
Figure 1. Western blot analysis and sequencing of 3 groups of structurally-distinct cells that may end up with the same results. (Source: BIORENDER)
Using single-cell sequencing, in which each cell is analyzed individually, it is possible to map cell lineage and detect heterogeneity between cells. Figure 2 shows a single-cell sequencing analysis of the immune microenvironment of breast tumors at Memorial Sloan Kettering Cancer Center and Columbia University, which generated an immune map of breast cancer, reflecting different stages of T-cell activation and differentiation.
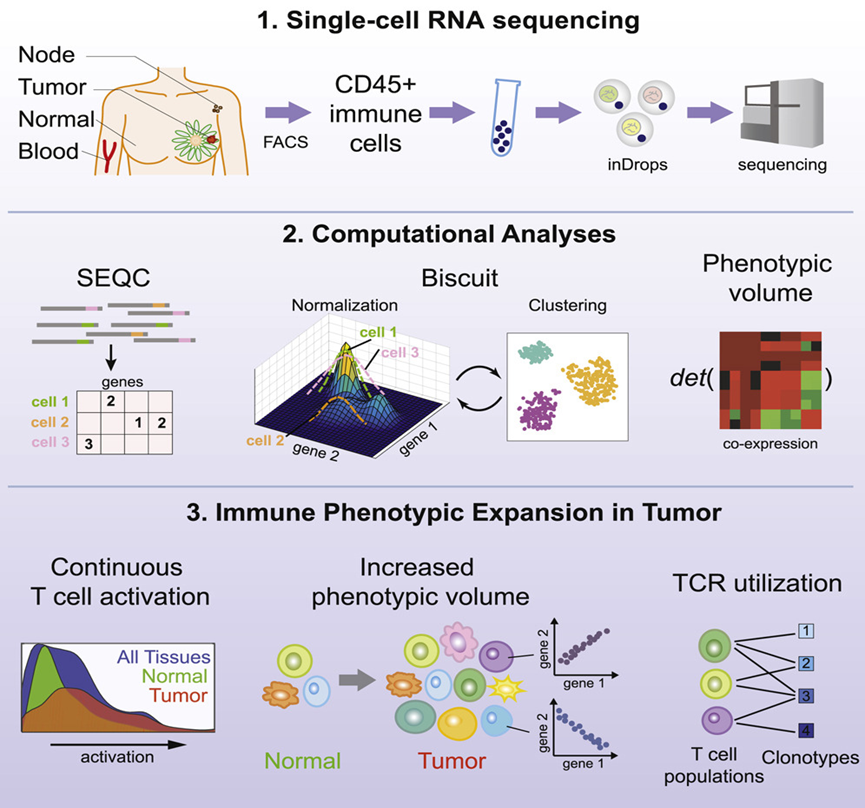
Figure 2. The technical roadmap of single-cell RNA sequencing. [1]
Application of Single-Cell Sequencing in Cancer Research
To capture the genetic heterogeneity of cancer cells, deep sequencing of large amounts of DNA extracted from tumor tissue is required. This approach is based on three factors: [2]
For some primary tumors that are difficult to biopsy, clinicians usually extract materials through fine-needle aspiration (FNA) for histopathological examination and diagnosis. However, FNA sometimes doesn't provide enough material for deep sequencing. Thus, the single-cell sequencing approach facilitates the analysis of such tumors.
In addition, the detection of clonal and subclonal somatic mutations with deep sequencing from cells in bulk requires accurate detection of the mutated DNA molecules in the data. In some tumors (e.g., pancreatic cancer), due to stromal infiltration, cancer cells only occupy a part of the sample, and the remaining part may be composed of stromal components, which requires extremely high sequencing coverage.
Extensive genomic and transcriptomic studies of tumors help reveal the genomic complexity of cancer. Single-cell sequencing is a powerful technology that can help analyze genetic and transcriptomic heterogeneity within tumors to guide more effective therapeutic interventions.
Advanced Technologies for Single-Cell Sequencing
|
Single-cell Sequencing |
Features |
Functions |
|
SCI-seq |
Single-cell combinatorial indexing |
Simultaneous construction of thousands of single-cell libraries and detection of somatic copy number changes |
|
LIANTI |
Single-cell whole-genome amplification |
Detection of cellular copy number variations and disease-associated mutations |
|
scCOOL-seq |
Single-cell multiplex sequencing |
Detection of chromatin state, nucleosome positioning, DNA methylation, copy number variation and ploidy |
|
TSCS |
Providing accurate spatial information |
Description of the spatial characteristics of individual tumor cells |
|
SiC-seq |
High Throughput, Low deviation |
Extensive study of the genomes of different cells |
|
Microwell-seq |
High throughput, low cost |
Enhancing assay abundance in single-cell sequencing |
|
SPLit-seq |
Combined with the principle of combinatorial barcoding labeling, low cost |
Single-cell transcriptome sequencing |
Limitations of Single-Cell Sequencing
Despite great progress in developing single-cell sequencing, significant limitations still remain. There are some problems associated with whole-genome amplification (WGA) on the DNA side, with examples including low genomic coverage and allelic loss (i.e., random, preferential amplification and sequencing of one of the two alleles of any given gene in the genome of a diploid single-cell). Also, there is a lack of methods capable of enumerating DNA molecules, which would greatly aid in the identification of allelic loss, especially in polyploid cancer genomes. In addition, both the inherent chemical instability of nucleic acids (e.g., deamination of cytosine bases) and the error rates associated with polymerases used to amplify single-cell DNA further increase the difficulty of sequencing.
As for RNA, it is difficult to attribute observed variation in single-cell data to technical deviation or biological variation because of data sparseness, variance of sample, and loss of gene expression for low-level transcripts. These limitations are largely due to the relatively low efficiency of mRNA capture (5%-15%) in single-cell transcriptomic approaches. [2]
Applications of Single-Cell Sequencing
Single-cell sequencing can be used to study the genetics and biology of various stages of cancer, such as subpopulations of cells capable of causing cancer, precancerous lesions, carcinoma in situ, cancer metastasis, and more. Cells in tumor tissue have different genetic and phenotypic characteristics from healthy tissues. By detecting the heterogeneity of tumor cells, the maps of tumor cells and the tumor microenvironment can be made, so as to further clarify the cell population in the tumor tissue, find specific markers, and interpret the tumorigenesis and metastasis.
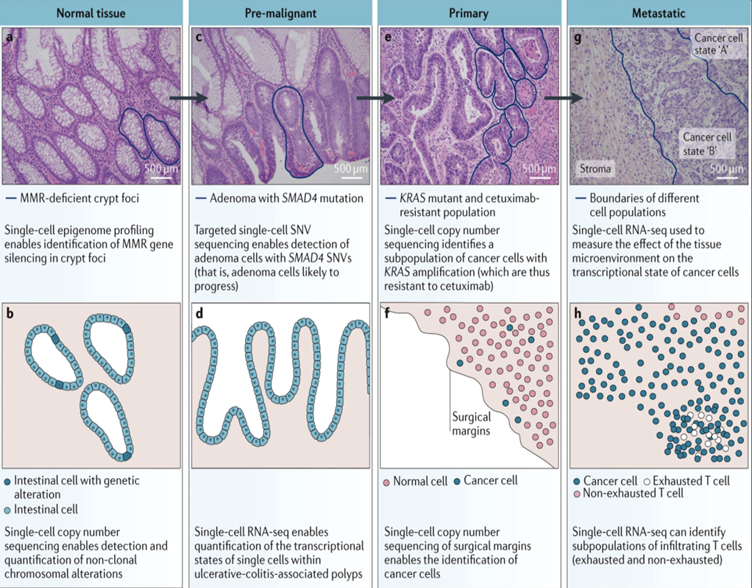
Figure 3. Diagram of hematoxylin and eosin (H&E) staining and different stages of colorectal adenocarcinoma, and the applications of single-cell sequencing at various stages. [1]
Application of Single-Cell Sequencing in CAR T-Cell Therapy
CAR T-cell therapy has made great progress in the treatment of hematological tumors. At present, 8 products related to CAR T-cell therapy have been launched, of which 6 are targeting CD19 antigen and 2 are targeting BCMA antigen. However, CAR T-cell therapy can cause a variety of adverse reactions, including neurotoxicity, and the mechanism of which is still unclear. Researchers from Stanford University, led by Kevin R. Parker and Denis Migliorini, discovered the expression of CD19 antigen in human brain parietal cells using single-cell sequencing, and speculated that the “on-target, off-tumor” attack of CAR T-cells on parietal cells may be the potential cause of neurotoxicity. [3]
The study first analyzed scRNA-Seq datasets to identify CD19-positive cells in humans and found that CD19 is highly expressed in human brain parietal cells.
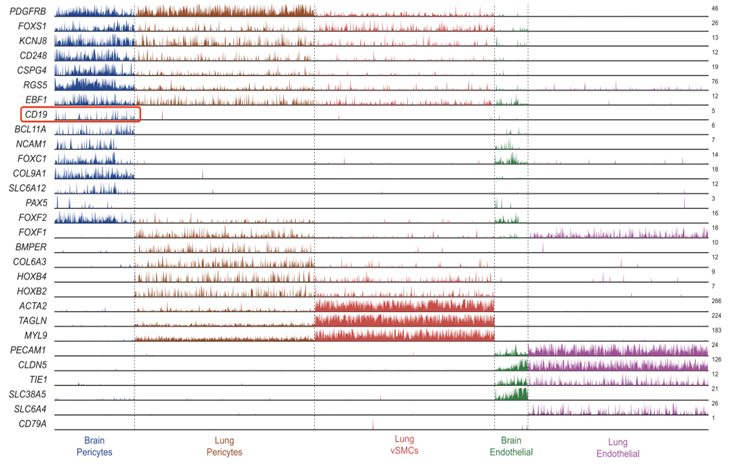
Figure 4. CD19 is specifically expressed in brain pericytes. [3]
The researchers then performed neurotoxicity assays in mouse models and found CD19-positive cells in the brain tissue of C57BL/6J mice. In order to avoid the influence of B-cells expressing CD19 antigen on experimental results, they used severe combined immunodeficiency (SCID) mice (NSG mice, no B cells), and constructed CAR T-cells to verify neurotoxicity. It was found that CAR T-cells targeting murine CD19 caused an increase in the permeability of the blood-brain barrier, while CAR T-cells targeting human CD19 had no such effect.
By comparing the single-cell sequencing results of mouse and human brain pericytes, it was found that the expression of CD19 in mouse brain pericytes was lower than that of human brain. Furthermore, there are specific differences between species, so the cell types and transcriptional status of mice may not fully correspond to human-specific pathophysiology. Initial studies of CAR T-cells in mouse models reportedly failed to predict the degree of neurotoxicity later observed in human clinical trials.
Therefore, the use of scRNA-seq to analyze the expression of related antigens by Kevin R. Parker and Denis Migliorini is extremely important for the study of CAR T-cells. It captures rare cell populations, and its indexes may be critical in detecting the clinical effects of targeted therapies. In the future, CAR T-cells will have more powerful functions and may recognize combinations of multiple target antigens, and will be increasingly applied in the research of solid tumors. Solid tumors share most antigens with normal tissues, so single-cell sequencing can be used to discover and design targets with high efficiency and specificity to tumor cells, making it a powerful tool for immunotherapy research.
Why Choose Cyagen?
In the preclinical research of CAR T-cell therapies, the selection of cell models, animal models, and modeling strategy is crucial. Clarifying the purpose of the research and establishing effective experimental models and protocols will make the process of biomedical translation more efficient.
Cyagen provides one-stop services to help CAR T-cell therapy research and development, including antibody screening, stable cell lines construction, development of cell/animal models, and drug efficacy evaluation. We also offer downstream model support, including colony management, breeding, phenotype analysis services, and more.
Request a quote now. Alternatively, you can always email service-apac@cyagen.com or call 86 20-31601779 to inquire about our services or obtain a quote for your project.
[1] Azizi, E., Carr, A. J., Plitas, G., Cornish, A. E., Konopacki, C., Prabhakaran, S., ... & Pe’er, D. (2018). Single-cell map of diverse immune phenotypes in the breast tumor microenvironment. Cell, 174(5), 1293-1308.
[2] Baslan, T., & Hicks, J. (2017). Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nature Reviews Cancer, 17(9), 557-569.
[3] Parker, K. R., Migliorini, D., Perkey, E., Yost, K. E., Bhaduri, A., Bagga, P., ... & Satpathy, A. T. (2020). Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell, 183(1), 126-142.
[4] Giavridis, T., van der Stegen, S. J., Eyquem, J., Hamieh, M., Piersigilli, A., & Sadelain, M. (2018). CAR T cell–induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nature medicine, 24(6), 731-738.
We will respond to you in 1-2 business days.