
There's an old saying that eyes are the windows to the soul. What if the windows were closed and you could no longer see the world clearly? Can you imagine yourself living in a world where there is only darkness? There is a group of such people who are suffering from the fear of going blind; they are patients living with Leber congenital amaurosis (LCA).
In June 2021, the first clinical study of gene therapy for LCA in China was officially launched in Shanghai. A few months later, the operation of 6 patients was successfully completed, and their visual function improved to varying degrees. The safety of RPE65 gene therapy preparations was confirmed. As soon as the news came out, it undoubtedly brought new expectations to this special group.
Leber Congenital Amaurosis (LCA)
Leber congenital amaurosis (LCA; OMIM 204000) is a rare inherited retinal dystrophy (IRD) that typically presents with early-onset, potentially blinding visual impairment. LCA accounts for more than 5% of all retinal diseases, and the current prevalence in the world is about 1:30000~1:81000. It also causes blindness in 10% to 20% of children afflicted, and is the most common cause of inheritable blindness in childhood. The main clinical features are severe visual impairment, nystagmus, fundus pigmentation, and severely abnormal electroretinogram within one year of birth, which may be accompanied by myopia or hyperopia, dactylopia, visual fixation disturbance, and neurological symptoms.
Clinical Features of LCA
LCA is highly clinically heterogeneous, with clinical phenotypes including retinal dysfunction (color blindness, night blindness), loss of central or peripheral vision, and complete blindness due to rod and cone damage. Patients with this disease may have symptoms of nystagmus after birth, and symptoms such as optic nerve atrophy, macular defect or atrophy, and osteocyte-like pigment migration in the fundus. In addition, patients may also experience symptoms of photophobia, night blindness, and intellectual disability.
The currently accepted clinical diagnostic criteria for the disease are 1) Low vision or loss of vision at birth or a few months after birth; 2) Sluggish or absent pupillary responses; 3) Nystagmus; 4) Eye-poking syndrome; 5) Retinal electrophysiological signals disappear; 6) Fundus abnormalities.
Eye-poking syndrome, which is a repetitive act of poking or pressing the eyeball with knuckles or fingers, is often seen in children with enophthalmos. The action of poking the eyeball can cause the eyeball to sink deep, so it may also be a prominent facial feature in people with LCA.
Most of the patients with LCA have relatively stable visual function. Some patients lose visual function, and very few patients can improve their visual function. Patients with this disease often have high hyperopia or high myopia. Visual function and visual acuity in patients with LCA vary greatly, ranging from no light to 20/200. Patients with mutations in the CRB1, LRAT and RPE65 genes have visual acuity ranging from no light to 20/50.
The disease causes various abnormalities in the fundus, such as typical retinitis pigmentosa, white retinal spots, macular defects, optic disc pseudocapillary lesions, and hyperpigmentation. Retinitis pigmentosa, or retinitis leukoplakia, is the most common fundus abnormality, usually seen in older children. A macular defect is a prominent retinal feature in LCA, however, this defect is not a developmental abnormality but a complete loss of foveal retinal tissue.
Keratoconus is a degenerative, non-inflammatory disease of the cornea characterized by thinning of the cornea, changes in the shape of the cornea to a cone shape, and changes in the radius of curvature. Keratoconus is mostly associated with LCA, which further impairs the patient's visual function, sometimes associated with cataracts.
Because the disease-causing genes of LCA CEP290 and IQCB encode ciliary proteins, these two mutations cause not only LCA but also complex ciliary disorders, including Joubert syndrome, Senior Loken syndrome (SLS), Bardet-Biedl syndrome, and Meckel-Gruber syndrome. These syndromes can affect the central nervous system (CNS) and other organs such as kidneys, liver, bones, or heart.
Disease-Causing Gene Mutations of LCA
LCA is hereditary, inherited mainly in an autosomal recessive manner. To date, more than 300 genes have been found to be associated with inherited retinal diseases, of which, 28 are responsible for different types of LCA. These genes have important functional roles in retina and pigment epithelium, mainly including guanine synthesis (IMPDH1), retinal differentiation (OTX2), photoreceptor cell morphogenesis (CRB1, CRX, and GDF6), ciliary transport processes (CEP290, CLUAP1, IFT140, IQCB1, LCA5, RPGRIP1, SPATA7, and TULP1), phototransduction (AIPL1, GUCY2D and RD3), retinoid cycle (LRAT, RDH12 and RPE65) and signal transduction (CABP4, KCNJ13).
|
Name |
Disease-Causing Gene |
Protein |
Protein Function |
|
LCA1 |
GUCY2D |
Guanylate cyclase 2D |
Phototransduction |
|
LCA2 |
RPE65 |
Retinoic acid isomerase |
Retinoid cycle |
|
LCA3 |
SPATA7 |
Spermatogenesis-related protein 7 |
Photoreceptor cilia transport |
|
LCA4 |
AIPLI |
Aryl-hydrocarbon-interacting-protein-like 1 |
Phototransduction/Protein biosynthesis |
|
LCA5 |
LCA5 |
Lebercillin |
Photoreceptor cilia transport |
|
LCA6 |
RPGRIPI |
Retinitis pigmentosa GTPase regulation-related protein 1 |
Photoreceptor cilia transport |
|
LCA7 |
CRX |
Cone-rod homeobox protein |
Photoreceptor morphogenesis |
|
LCA8 |
CRBI |
Crumbs homologue 1 |
Photoreceptor morphogenesis |
|
LCA9 |
NANAT |
Nicotinamide nucleotide adenosyltransferase 1 |
Coenzyme NAD biosynthesis |
|
LCA10 |
CEP290 |
Centrosome protein 290 |
Photoreceptor cilia transport |
|
LCA11 |
IMPDHI |
Inosine 5-monophosphate dehydrogenase 1 |
Guanine synthesis |
|
LCA12 |
RD3 |
Protein RD3 |
Protein transport |
|
LCA13 |
RDH12 |
Retinol dehydrogenase 12 |
Retinoid cycle |
|
LCA14 |
LRAT |
Lecithin retinol acyltransferase |
Retinoid cycle |
|
LCA15 |
TUP1 |
Tubular protein |
Photoreceptor cilia transport |
|
LCA16 |
KCNJ13 |
Kir7 inward rectifier potassium channel |
Phototransduction |
|
LCA17 |
GDF6 |
Growth differentiation factor 6 |
Photoreceptor morphogenesis |
LCA is mainly caused by loss of function mutations in important genes that affect retinal function. LCA can be divided into as many as 17 types based on the chromosome number of the variant and the location of the specific affected gene. The most common of these are Leber congenital amaurosis 2 (LCA2) and Leber congenital amaurosis 10 (LCA10), which are caused by mutations in the RPE65 gene on chromosome 1 and CEP290 on chromosome 12, respectively.
Along with in-depth research on ophthalmic diseases, the development of related animal models has also made major breakthroughs in ophthalmic disease research. At present, for LCA2, LCA10, retinitis pigmentosa (RP), retinal degeneration, macular degeneration, corneal endothelial dystrophy and other ophthalmic diseases, Cyagen can provide or develop gene edited mouse models, such as knockout, knock-in, point mutation, humanized mouse models, and surgical rodent models, to accelerate the development of pharmacodynamic verification experiments.
Advances in Gene Therapy Research for LCA
There are two main gene therapy drugs for LCA, which are for LCA2 and LCA10 respectively.
The gene therapy product for LCA2 is called Luxturna, developed by Spark Therapeutic and launched in the United States in 2017. The drug, which costs US$430,000 for a single eye, is the first gene therapy in the world to use AAV delivery to treat retinal diseases. The method restores retinal function in patients by delivering functional RPE65 protein to retinal pigment epithelial cells, requiring regular injections.
The gene therapy product for LCA10 is EDIT-101, a potential treatment developed by Editas Medicine that still undergoing clinical trials. The drug uses AAV-mediated delivery of Targeted Gene Editing to target both upstream and downstream of the mutated region through sgRNA, so as to delete or invert the mutated region to treat LCA10. Notably, the therapy can cure the disease with a single administration.
Due to the large number of LCA types, the lack of research data, and the difficulty in patient recruitment, there has been a lack of effective treatments for LCA. The above two gene therapy products bring the hope of a complete cure for the disease. However, because it can only target two types of LCA, it cannot solve the difficulty that most patients have no drugs available.
Obtaining a large amount of information related to clinical research is of great benefit to overcome these types of inherited retinal ophthalmic diseases. The Rare Disease Data Center (RDDC), jointly developed by the Pearl River Delta Research Institute of Tsinghua University and Cyagen, cooperates with all parties to break down data barriers and make clinical diagnosis and treatment more accurate and efficient.
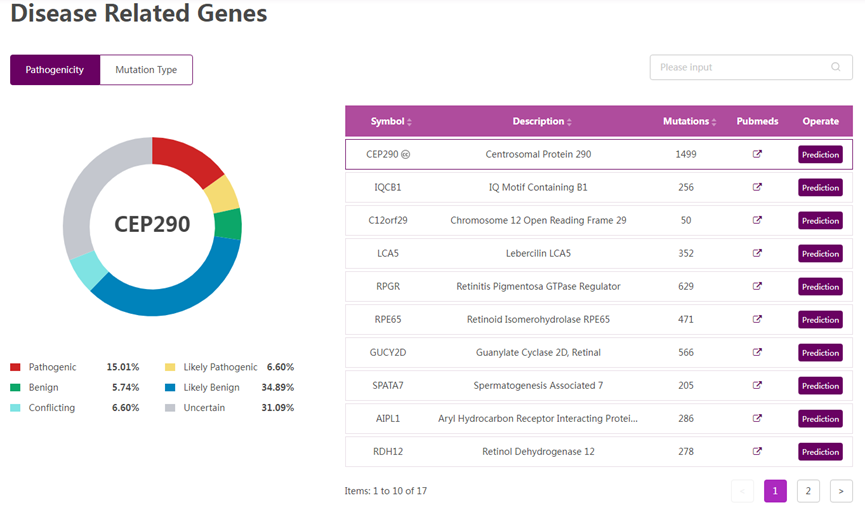
Figure 1 Disease-related information of LCA10. (Source: The Rare Disease Data Center)
Searching for Leber congenital amaurosis type 10 (LCA10) in RDDC can provide disease phenotype, disease-related information, target drugs, clinical trials, animal models, and more.
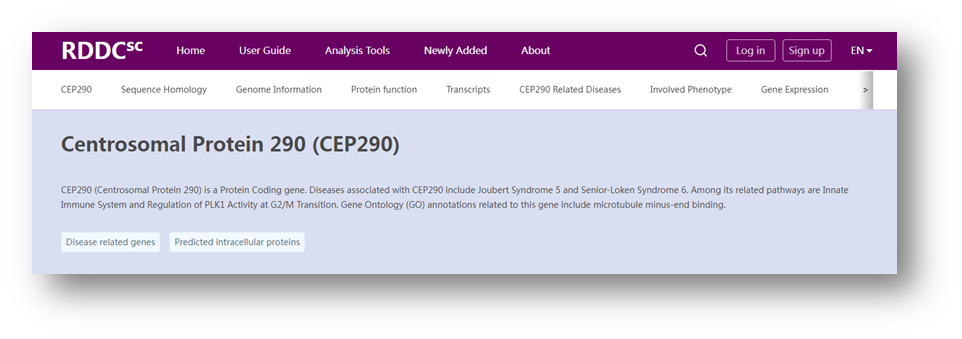
Figure 2 Related information of CEP290. (Source: The Rare Disease Data Center)
Searching for the disease-causing gene CEP290 of Leber congenital amaurosis type 10 (LCA10) in RDDC can yield the gene sequence homology, genomic information, clinical mutations, transcripts, related diseases, phenotypes, and gene expression levels.
Preclinical Ophthalmology Research Solutions
As a comprehensive contract research organization (CRO) solution provider, Cyagen recognizes ophthalmic diseases as a breakthrough point for gene therapy and has established an ophthalmic gene therapy platform to overcome the above obstacles. We have equipped the platform with state-of-the-art ophthalmic instruments for small animals and an experienced professional team.
With 16 years of gene editing model construction experience, Cyagen is ready to provide you with an array of standardized preclinical research solutions for ophthalmic gene therapy. If you have questions or need help with your ophthalmology drug development program, you’re invited to have a complimentary talk with our experts!
We will respond to you in 1-2 business days.