
On March 4, 2022, the Beijing Winter Paralympic Games were grandly initiated at the National Stadium. At the opening ceremony, Chunyan Wang, who was wearing a blue gauze skirt, sang the theme song of the Winter Paralympic Games "In the Beautiful Snow" with a clear and melodious voice in the center of the stage. If you only look at her appearance, no one would have thought that this girl with bright eyes is living with a disability.
Chunyan Wang has a rare disease called retinitis pigmentosa (RP). After being diagnosed at the age of 4, her world gradually became irreversibly blurred, until she could only get a faint sense of light. Another celebrity with RP is Liwen Cai, a swimming champion at the 2020 Tokyo Paralympic Games.
Retinitis pigmentosa (RP) is a disease caused by a genetic mutation for which there is currently no effective treatment. The initial retinal degenerative symptoms of RP are decreased night vision (nyctalopia), the loss of the mid-peripheral visual field, and then decreased visual acuity until complete blindness occurs.

Figure 1. Chunyan Wang (left) and Liwen Cai (right).
Disease Description
Retinitis pigmentosa (RP) is a genetic disorder of the eyes that causes loss of vision.
The average global prevalence is approximately 1:4000, although there are some notable regional variations. In the Jerusalem area, the prevalence of non-syndromic RP is 1:2086, which is 2.5 times higher than in the United States and Europe. In China, the prevalence is about 1:10000, and it reaches 1:1000 among the elderly in northern China.
RP can be divided into syndromic RP and non-syndromic RP:
Syndromic RP involves multiple organs, and there are about 30 RP-related syndromes, including Usher syndrome (RP combined with deafness), Bardet–Biedl syndrome (RP with obesity, cognitive impairment, polydactyly, hypogonadism, renal insufficiency), metabolic diseases (methylmalonic aciduria, homocystinuria, abetalipoproteinemia, Bietti crystal-like retinal pigment epithelial degeneration, cystinosis), and nervous system diseases (myotonic dystrophy, Hallervorden-Spatz syndrome).
Non-syndromic RP(RP appears alone without other co-morbidities), accounting for 70% to 80% of all cases, usually presents a variety of the following clinical symptoms: night blindness, eventual blindness, cataract, bone spicule pigmentation in the fundus and more.
Epidemiological Study
RP is highly genetically heterogeneous, and inheritance patterns include: autosomal dominant (ADRP), autosomal recessive (ARRP), X-linked (XLRP), digenic (controlled by two genes) and mitochondrial. Among them, ADRP accounts for 30% to 40%, ARRP for 50% to 60%, and XLRP for 5% to 15%.
In non-syndromic RP, ADRP is usually the mildest type. The average age of onset is 20 to 30 years old, and some develop after the age of 50. Therefore, ADRP should be considered in the diagnosis of patients with mild symptoms and in older age. ARRP tends to develop in adolescence and is more severe than ADRP, but milder than XLRP. XLRP patients are all male, and it is transmitted across generations. Patients have an early onset of symptoms, usually within 10 years of age. The disease progresses rapidly, the condition is severe, and it is often complicated by high myopia.
Stages of Disease Progression
In the early stage of RP, symptoms are mainly mild night blindness, which may last for several years. Peripheral vision defect may occur in dim light, but may be mild or disappear in normal light. At this stage, the condition is relatively stable, and the symptoms do not affect the normal life of the patient, so it is difficult to make an accurate diagnosis. As shown in Figure 2, there is no or rare bone spicule-shaped pigment deposits was found in the fundus imaging examination in the early stage of the disease, the retinal arterioles were not constricted, and the optic disc was normal.
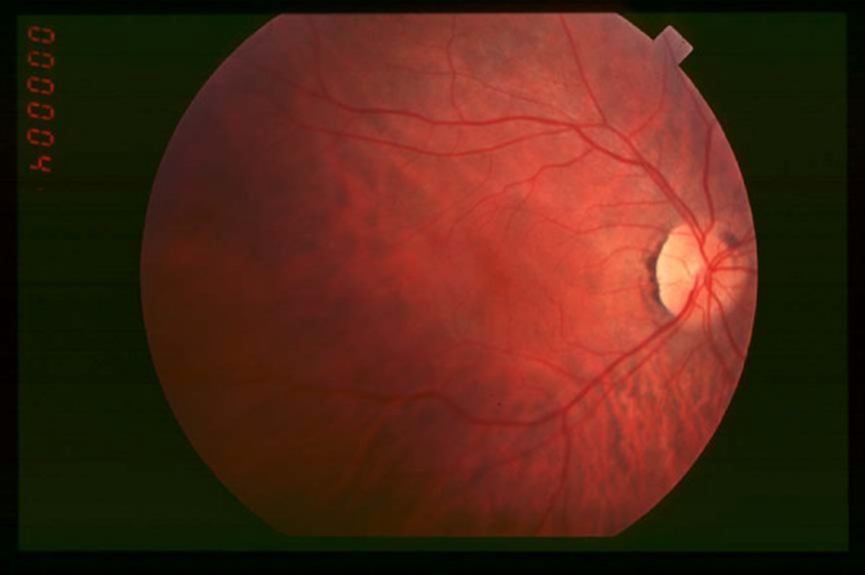
Figure 2. Fundus imaging in the early stage of RP.
In the middle stage of RP, the night blindness is aggravated, and the patient cannot travel normally in a dim environment. In a well-lit environment, the patient still has symptoms of peripheral vision defects and abnormal color vision. The retina may develop macular edema or mild foveal atrophy. As shown in Figure 3, bone spicule-shaped pigment deposits and atrophy of the retina were evident in the fundus imaging examination in the middle stage of the disease.

Figure 3. Fundus imaging in the middle stage of RP.
In the late stage of RP, due to the loss of peripheral vision, the patient's visual ability is greatly reduced. The patient needs a magnifying glass to assist vision, and has a strong photophobia, which greatly affects his/her quality of life. As shown in Figure 4, extensive pigment deposits appeared in the late fundus imaging examination, even reaching the macular region, the optic disc is pale, and the retinal arterioles are constricted. At this stage, fluorescein angiography can detect chorioretinal atrophy in the peripheral and foveal regions.
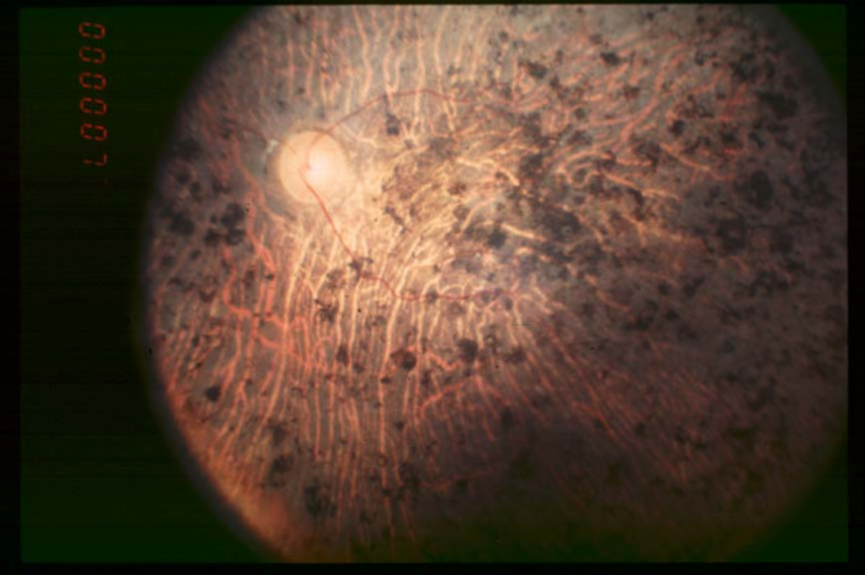
Figure 4. Fundus imaging in the late stage of RP.
Diagnostic Methods
1. Dark adaptation and color vision examination. Abnormal dark adaptation threshold is another characteristic of RP. This is due to reduced rod sensitivity and prolonged sensitivity recovery time, resulting in an increased rod threshold. Color vision may be normal in the initial stage of the disease, but color vision disorders, especially blue-yellow color vision disorders, develop in later stages.
2. Visual field examination. One of the hallmarks of RP is the progressive loss of visual field with high bilateral symmetry, usually starting with isolated scotomas in the mid-peripheral region and gradually coalescing to form partial or complete annular scotomas. As the disease progresses, the annular scotoma extends inward and outward (inward slowly). In addition, concentric visual field loss without annular scotoma, and visual field defects that progress in an arc from above to below the retina are also reported. Kinetic perimetry is best for assessing peripheral visual field loss, and the progression of central visual field loss is usually assessed using static perimetry.
3. Electroretinogram (ERG). ERG abnormalities occur in the early stage, and the scotopic ERG (cone and rod mixed reaction) shows that the generated wave is lower than normal. In full-field ERG examinations, rod cells were delayed, attenuated, or disappeared in response to dark flashes in dark-adapted examinations. As the disease progresses, full-field ERG may fail to record despite residual fields.
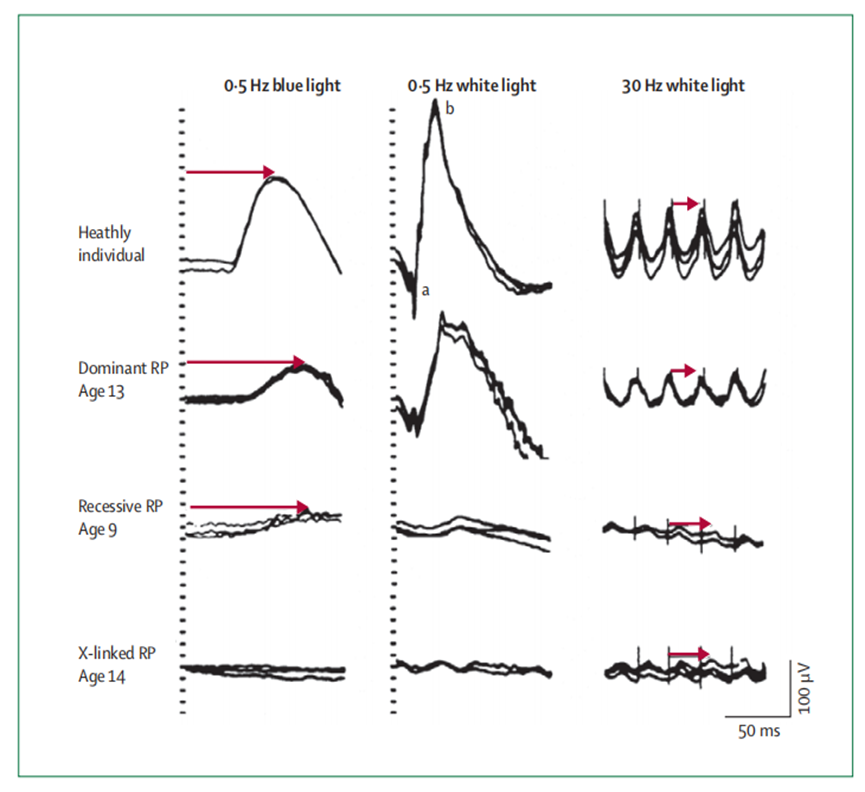
Figure 5 ERG responses in healthy individuals and three patients with early-stage RP, inherited in an autosomal dominant, autosomal recessive, and X-linked pattern, respectively.
4. Fundus photography. The visual field of traditional fundus photography is small, only 30-50 degrees, and is limited by turbid media, insufficient pupil dilation, and patient cooperation. There is now an option for ultra-wide-angle fundus photography, which can capture up to 200 degrees of retinal coverage in a single shot. However, this technique has the disadvantage of poor pixels, and preretinal structures such as eyelashes and vitreous opacities can create artefacts that interfere with diagnosis. Color imaging can use the reflectivity of three specific wavelengths of laser light to provide information at different levels of the retina. For the examination of patients with RP, color imaging can better define the macular boundary than traditional fundus photography.
5. Optical coherence tomography (OCT). The earliest histopathological change in RP is shortening of the outer segment of photoreceptor cells, which is reflected in frequency-domain OCT images as a disorder of the outer retinal layers. As RP progresses, the thickness of the outer segment and outer nuclear layer of photoreceptor cells decreases, to the complete loss of the photoreceptor outer segment and outer nuclear layer at late stages.
RDDC Powers Your Rare Disease Research

The Rare Diseases Data Center (RDDC) was developed by Research Institute of Tsinghua, Pearl River Delta (RITPRD) and assisted by Cyagen.
RDDC has collected and sorted out data and information about genes, mutations, phenotypes and animal models of various types of rare diseases from international public biological resources, aiming to present them in a visual way. At present, it has collected information on over 20,000 genes and 14,000 diseases!
Users can not only better understand the information of corresponding rare diseases, but also can take advantage of various AI tools to predict pathogenicity and RNA splice patterns, so as to help speed up the diagnosis and development of treatment research of rare diseases.
Preclinical Ophthalmology Research Solutions
As a comprehensive contract research organization (CRO) solution provider, Cyagen recognizes ophthalmic diseases as a breakthrough point for gene therapy and has established an ophthalmic gene therapy platform to overcome the obstacles faced by researchers. We have equipped the platform with state-of-the-art ophthalmic instruments for small animals and an experienced professional team.
With 16 years of gene editing model construction experience, Cyagen is ready to provide you with an array of standardized and custom preclinical research solutions for ophthalmic gene therapy. If you have questions or need help with your ophthalmology drug development program, you’re invited to have a complimentary talk with our experts!
[1] Zhang, Q. "Retinitis Pigmentosa: Progress and Perspective." Asia Pac J Ophthalmol 5 (2016).
[2] Verbakel, Sanne K., et al. "Non-syndromic retinitis pigmentosa." Progress in Retinal and Eye Research (2018).
We will respond to you in 1-2 business days.