
With unprecedented speed and influence, the golden age of gene therapy is coming! In recent years, several gene therapy drugs have been approved for marketing around the world, and many more are advancing through clinical trials. Gene therapy has given many patients with congenital diseases new hope! With over 100 types of monogenic inherited cardiovascular disease (CVD) discovered so far, the application of gene therapy has become a focus for recent CVD research and clinical trials.
The birth of a gene therapy drug is based on performing sufficient basic research with both innovation and integrity to be used for pre-clinical research and clinical trials. Today, we discuss a research paper published by a client of Cyagen Biosciences as an example to show you the foundational ideas of successful preclinical research on gene therapy for cardiovascular disease.

Background
Cardiac hypertrophy is an early sign of cardiovascular disease, which may subsequently lead to diastolic dysfunction, arrhythmias, and eventually heart failure. Therefore, understanding the key molecular mechanisms of pathological hypertrophy is crucial for the development of new therapeutic targets and strategies.
Researchers from the Cancer Hospital Affiliated with Harbin Medical University found that type III transforming growth factor-β receptor (TβRIII) plays an important regulatory role in the pathogenesis of cardiac hypertrophy. TβRIII induces cardiac hypertrophy through the β-arrestin2–dependent activation of CaMKII. The researchers used AAV9 vector-mediated specific shRNA to reduce the expression of TβRIII and found that the symptoms associated with cardiac hypertrophy were effectively alleviated. This study contributes to the development of novel therapeutic targets and strategies for the control of cardiac hypertrophy.
Foundational Research Ideas
The research publication was based on the following findings and methods, which integrated human and mouse biology to obtain the results discussed:
1. Abnormal expression of TβRIII was found in human cardiac hypertrophic tissue;
2. A TβRIII transgenic mouse model was constructed, and it was found that overexpression of TβRIII can induce cardiac hypertrophy in mice;
3. The expression of TβRIII was reduced by injection of AAV9-shTβRIII, followed by induction of cardiac hypertrophy by isoproterenol (ISO) infusion or transverse aortic constriction (TAC). It was found that the decreased expression of TβRIII can alleviate symptoms such as myocardial hypertrophy and cardiac function damage;
4. Mechanical study: it was found that TβRIII induces cardiac hypertrophy through activation of CaMKII by β-Arrestin2.
Results
The authors initially examined TβRIII expression profiles in human cardiac hypertrophic specimens. Significantly elevated expression of TβRIII was found in human cardiac hypertrophy samples, as were biomarkers such as atrial natriuretic peptide, brain natriuretic peptide, and β-MHC (Figure 1A, 1B, and 1C). Then myocardial hypertrophy was induced in mice by ISO infusion or TAC, and it was found that the expression of TβRIII in the left ventricle (LV) was significantly increased (Figure 1D and 1E). Moreover, immunofluorescence staining clearly showed that TβRIII was expressed in mouse LV tissue (Figure 1F, a). Line-scanning analysis of the fluorescence intensity suggested that TβRIII was mainly present in the vicinity of the plasma membrane of mature cardiomyocytes (Figure 1F, b). These results suggested that an increased expression of TβRIII may play a role in the development of cardiac hypertrophy.
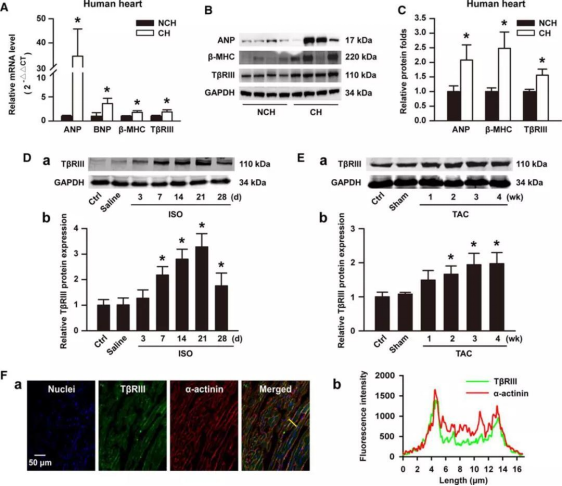
Figure 1. Increased expression of TβRIII in cardiac hypertrophic samples.
The researchers reproduced phenotypes (such as cardiac hypertrophy) in animal models by generating cardiac-specific TβRIII transgenic (CS-TβRIII-Tg) mice. They used the cardiac-specific α-MHC promoter to overexpress TβRIII in the heart of mice (Figure 2A) and demonstrated the successful establishment of the mouse model by Western blotting (WB) and quantitative polymerase chain reaction (qPCR) (Figure 2B and 2C) . Then, a series of experiments were carried out using the CS-TβRIII-Tg mouse model established by Cyagen Biosciences.
Relative to wild-type (WT) mice, the levels of atrial natriuretic peptide, brain natriuretic peptide and β-MHC proteins were increased in CS-TβRIII-Tg mice (Figure 2 D). Increased LV wall thickness and LV mass were detected in CS-TβRIII-Tg mice using serial echocardiography (Figure 2E). Comparative analysis of histology, ratios of heart weight/body weight, and heart weight/tibia length revealed that cardiac-specific overexpression of TβRIII resulted in spontaneous cardiac hypertrophy (Figure 2F, 2G, and 2H). In addition, treated with ISO or TAC, the degree of cardiac hypertrophy was more severe in CS-TβRIII-Tg mice than in wild-type mice (ISO: Figures 2I through 2K; TAC: Figures 2L through 2N). To sum up, cardiac overexpression of TβRIII was sufficient to induce cardiac hypertrophy, and that elevated expression of TβRIII accelerated stress-induced injury of heart function.
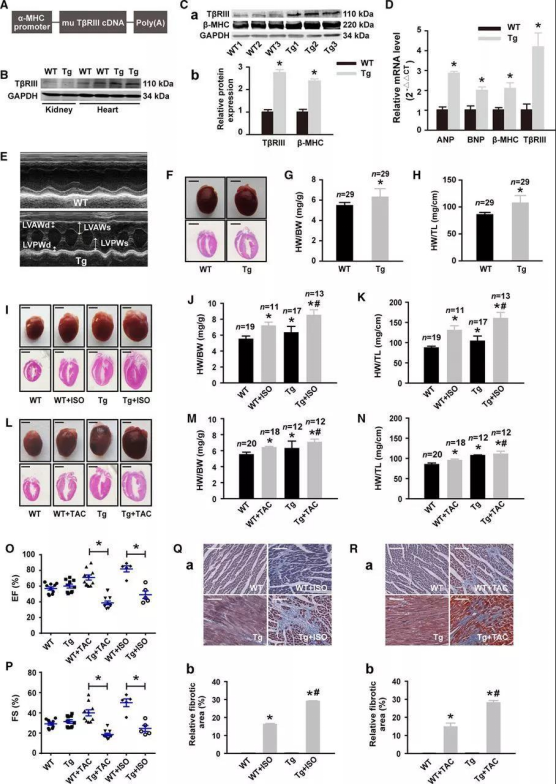
Figure 2. Cardiac-specific overexpression of TβRIII promotes cardiac hypertrophy.
The authors speculated: since overexpression of TβRIII can promote cardiac hypertrophy, could reducing the expression of TβRIII alleviate stress-induced cardiac hypertrophy? They used cardiac-targeted AAV9 vector-mediated specific shRNA (AAV9-shTβRIII) to knockdown TβRIII gene in mouse hearts and verified the phenotype by WB (Figure 3B).
Compared with WT mice, the hypertrophic index induced by ISO or TAC treatment in shTβRIII mice was significantly reduced, and myocardial hypertrophy and cardiac function impairment were alleviated. (Figure 3C). By calculating the ratios of LV mass/body weight and LV mass/tibia length, it was also demonstrated that the knockdown of TβRIII reduced the cardiac hypertrophy indices (Figure 3D through 3G). Furthermore, 12 weeks after TAC operation, the ejection fraction (EF) and fractional shortening (FS) declined remarkably in both the WT and the AAV9-mediated scramble shRNA-infected mice, which indicates that reducing the expression of TβRIII can alleviate the myocardial contractile function damage caused by TAC (Figure 3H and 3I). Furthermore, the increased expression levels of cardiac hypertrophy markers β-MHC protein and atrial natriuretic peptide, brain natriuretic peptide, and β-MHC mRNA induced by ISO or TAC were significantly decreased by the cardiac-specific knockdown of TβRIII (Figures 3J through 3L).
In this study, the researchers used AAV9 vector-mediated specific shRNA to reduce the expression of endogenous TβRIII in mice and observed that shTβRIII mice had a significantly reduced cardiac hypertrophic disease phenotype compared to control mice. This indicates that reducing the expression level of TβRIII can improve cardiac function by alleviating damage, and also verifies the function of TβRIII in the pathogenic mechanism of cardiac hypertrophy.
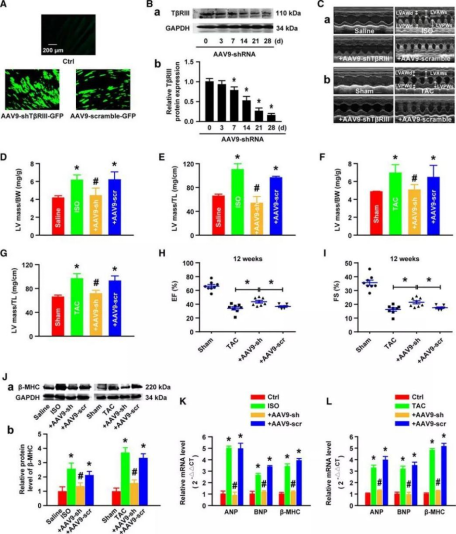
Figure 3. Reducing the expression of TβRIII alleviates cardiac hypertrophy.
After analysis, the researchers found that TβRIII can interact with β-Arrestin2 to affect proliferation and migration in various cancer cell types and that calmodulin-dependent protein kinase II (CaMKII) plays an important role in this process. Therefore, the authors conducted in vivo and in vitro studies and finally found that TβRIII interacts with CaMKII through β-Arrestin2 to induce cardiac hypertrophy.
Conclusions
In this study, the authors provide substantial and accurate data showing that TβRIII induces cardiac hypertrophy through β-Arrestin2-dependent CaMKII activation. This finding contributes to further understanding of the mechanism of cardiac hypertrophy and confirms that interfering with TβRIII expression in cardiomyocytes by the AAV delivery system may be a way to prevent and cure cardiac hypertrophy.
Why Choose Cyagen?
As a comprehensive solution provider, Cyagen has established an innovative CRO platform that provides researchers with leading-edge genetically modified model services. We provide one-stop solutions for gene therapy development to meet the needs of all levels of research. Our services include Targeted Gene Editing Targeted Gene Editing Screening & Target Validation, Custom Mouse Model Generation, Efficient Adenovirus/Lentivirus Packaging of adeno-associated virus (AAV), lentivirus (LV), adenovirus (ADV), Pharmacology and Pharmacodynamics (PD) Studies, and more. Our services aim to improve the efficiency of gene therapy research results for investigators worldwide.

Lou, Jie, et al. “Type III Transforming Growth Factor-β Receptor Drives Cardiac Hypertrophy Through β-Arrestin2-Dependent Activation of Calmodulin-Dependent Protein Kinase II.” Hypertension, vol. 68, no. 3, 2016, pp. 654–666.
We will respond to you in 1-2 business days.