
Huntington's disease (HD), also known as Huntington's chorea, is a neurodegenerative disease that is mostly inherited. In our previous article on Huntington's disease, we covered the symptoms, pathological changes, and treatment of the disease. In this article, we will further demystify HD, introducing its causative genes, other treatments, and the application of related animal models.
HD-Causing Gene Mutation
Huntington's disease (HD) is caused by a mutated form of the huntingtin gene (HTT), where excessive (more than 36) CAG repeats result in the formation of an unstable HTT protein. In a normal person, the number of CAG repeats is between seven and 35 repeats. People with 36 to 40 CAG repeats may or may not develop the signs and symptoms of Huntington's disease, while people with more than 40 repeats will develop the disorder during a normal lifetime. It should be noted that the onset time of HD is related to the number of CAG repeats, and the more repeats, the earlier the onset. Dyskinesia usually begins at age 40 in those with about 40 CAG repeats and begins in adolescence in those with more than 60 CAG repeats.
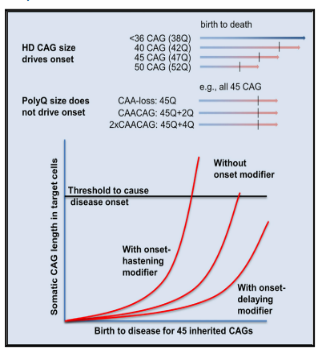
Figure 1. The number of CAG repeats determines the age of onset of HD.
The genetic causative factors of Huntington's disease have been clarified, but the pathological mechanism has not yet been fully and clearly reported. A relatively accepted statement is that mutant HTT protein accumulates and deposits in the striatum of the brain, and then damages neurons in the striatum, leading to neuronal apoptosis and striatal atrophy. When the HTT gene is mutated, the expression of mutant HTT can produce a variety of toxic variants, including RNA hairpins formed by small Cajal body-specific RNAs (scaRNA), abnormal repeat-associated non-ATG (RAN) translation protein products, N-terminal HTT gene exon 1 protein fragment formed by abnormal splicing and a smaller fragment of mutant HTT (mHTT) produced by further processing.
These products can either remain monomers or form oligomers or inclusion bodies, which may contribute to the pathogenesis of HD to varying degrees.
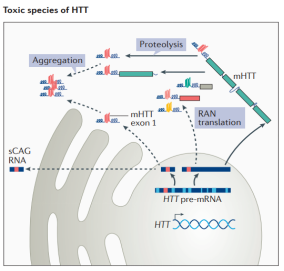
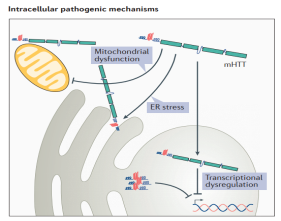
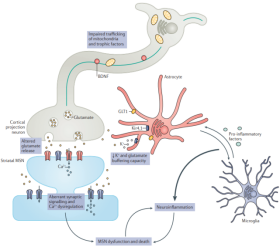
Figure 2. Multiple toxic variants of HTT contribute to the development of HD.
In HD, the presence of mHTT results in transcriptional dysregulation, impaired mitochondrial function, synaptic dysfunction, endoplasmic reticulum (ER) stress, and loss of trophic support for medium spiny neurons (MSNs). mHTT can impair extracellular ion homeostasis and reduce glutamate uptake by astrocytes at synapses by altering the expression of the inwardly rectifying K+ channel 4.1 and glutamate transporter 1 (GLT1), thus increasing the excitability and activation of neurons. In addition, mHTT can also cause abnormal immune activation of peripheral and central nervous system cells, leading to neuroinflammation, and ultimately neurodegeneration and neuronal apoptosis.
Research & Therapy
Gene therapy for HD is a key research direction of major research institutions and pharmaceutical companies in recent years. There are various methods of gene therapy, such as AAV vector delivery, antisense oligonucleotides (ASO) drugs, RNA interference (RNAi) and more.
The following are some of the gene therapy drug development projects currently in the clinical stage:
Boxun Lu's team at Fudan University has worked out an innovative method of drug discovery: using the autophagosome-tethering compounds (ATTEC) to degrade pathogenic proteins and treat the disease. They developed a "small molecule glue" that can specifically bind to the HD-causing protein mHTT and uses autophagy to reduce mHTT levels without affecting wild-type HTT levels. Studies have shown that the drug is able to cross the blood-brain barrier, and a small dose of intraperitoneal injection would significantly reduce mHTT levels in the cortex and striatum of HD mice. Such drugs can improve disease-related phenotypes without affecting wild-type HTT levels, providing an entry point for the development of oral or injectable drugs for HD.
Animal Models of HD
HD mouse models are mainly divided into two categories:
One is to overexpress the N-terminal fragment of mHTT in mice by transgenic technology, including the R6/2 and R6/1 models expressing the corresponding protein fragments of mutant HTT exon1, and the N171-82Q model expressing longer fragments. These N-terminal fragments, which all contain the polyQ sequence in mHTT, were also found to be highly toxic and prone to aggregation. This type of model is genetically inconsistent with HD patients because it only has the 5-terminal sequence of the mutant gene and often has multiple copies; and the phenotype often appears several weeks earlier, which is also inconsistent with most patients.
The other is to express full-length mHTT by two means. One is to express the transgenic human mHTT gene by artificial chromosome method, mainly including two models constructed by using yeast artificial chromosomes (YAC) and bacterial artificial chromosomes (BAC). The advantage of this method is that the human gene is expressed and contains all relevant elements of the gene in the human genome, including genomic sequences such as promoters and introns. The disadvantage is that it still belongs to the genetically modified model. Although the number of copies of the mutant gene was increased, the number of wild-type copies was not reduced, which was inconsistent with the patient's genotype. The other is to replace exon 1 in the mouse Htt gene with exon 1 in the patient's HTT gene to express mHTT. According to the number of CAG repeats, the commonly used mouse models include Q140, Q150, and Q175 models. These models are constructed by gene knock-in technology, using the mouse endogenous Htt promoter to express the mHTT protein, and replacing the original wild type, which is the closest genetically to HD patients. However, this method is also flawed. Except for exon 1 and some intron 1, the mutant genes expressed are all mouse homologous gene Htt, which has species differences.
In general, the advantages of mouse models for HD research lie in comprehensive research tools, and high genotype and phenotype matching with patients, which can provide key information for the study of HD's mechanism and treatment.
Scientists have also established a series of large animal models for studying HD:
One is the HD monkey model, which expresses the gene exon1 carrying 84 CAG repeats by lentivirus injection into egg cells. Prof. Xiaojiang Li and Prof. Shihua Li, who collaborated with Anthony Chan of Yerkes National Primate Research Center, reported in 2008 that they established an HD rhesus monkey model and successfully developed phenotypes such as dystonia and chorea.
The second is the HD sheep model, using the microinjection to transfer HTT full-length cDNA carrying 73 CAG repeats into fertilized eggs. The HD sheep model constructed by this method showed some HD pathological changes and metabolic phenotypes at the molecular level, but no obvious disease phenotypes in terms of nervous system function were observed.
The third is the very promising HD porcine model. Why is the porcine model so promising? Because the body size, phylogeny, and metabolism of pigs are very similar to those of humans, the size of the brain is also relatively close, and it has the same sulcus as humans. Therefore, porcine brain disease models have unique advantages and are more easily applied to clinical trials. The earliest HD porcine model was constructed by transgenic overexpression of the N-terminal fragment of mHTT. However, due to the high toxicity of expressing exogenous pathogenic gene fragments, the survival time is so short that the pathogenic gene cannot be passaged. To simulate neurodegenerative diseases more accurately, the researchers used CRISPR/Cas9 gene editing technology to insert the human mutant HTT gene into the endogenous gene of pigs and established the HD knock-in porcine model through somatic cell nuclear transfer. It does not introduce exogenous transgenes and directly changes the endogenous wild-type gene into the disease type so that the HD genotype is completely consistent with the patients. Further research found that this model could well simulate the typical pathological features of selective death of spiny neurons in the striatum of HD patients, and also showed HD-like phenotypes such as weight loss and abnormal behavior. More importantly, the above phenotypes can be passed onto future generations, providing a stable source of large animal models for HD drug development.
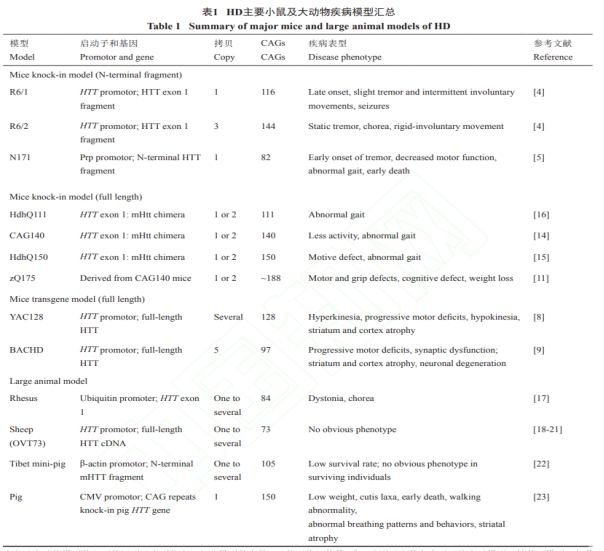
Figure 3. Summary of the main animal models of HD.
In addition to the above-mentioned animal models, there are also yeast models, nematode models, Drosophila models, zebrafish models, and more. These models also have specific advantages, such as high experimental throughput for phenotype detection, short turnaround times, and ease of high-throughput screening.
RDDC Facilitates Rare Disease Research

Log in to the official website (rddc.tsinghua-gd.org) for more information!
*STATEMENT: The data and tools in RDDC are for scientific research only, for reference only, and cannot be used as the conclusion of medical diagnosis and evaluation.
[1] https://en.wikipedia.org/wiki/Huntingtin#Gene.
[2] Zhaoyang L, Cen W, Ziying W, et al. Allele-selective lowering of mutant HTT protein by HTT-LC3 linker compounds. Nature. 2019 Nov;575(7781):203-209.
[3] Ping A, Boxun L. Current Research Status of Huntington's Disease. Chinese Journal of Cell Biology. 2018, 40(10): 1621–1632
We will respond to you in 1-2 business days.