
Huntington's disease (HD), also known as Huntington's chorea, is a neurodegenerative disease that has a 50% probability of being inherited by children of people living with HD. The earliest known description of the disease was made in 1841 by American physician Charles Oscar Waters. The condition was described in further detail in 1872 by American physician George Huntington. HD is an autosomal dominant degenerative disease of the basal ganglia and cerebral cortex, clinically characterized by insidious onset of symptoms including chorea (jerky movements), cognitive disorder, as well as mental and behavior disorder. As the disease advances, the uncoordinated, involuntary body movements of chorea become more apparent. Physical abilities gradually worsen until coordinated movement becomes difficult and the person is unable to talk. Mental abilities generally decline into dementia.

The age of onset ranges from childhood up to 79 years old, but usually begins between 30 and 50 years of age. Those diagnosed before the age of 20 are diagnosed to have juvenile HD, but they account for less than 10% of all HD cases. The symptoms of juvenile HD usually begin as chorea, and as the disease progresses, the patient gradually loses the ability to speak, move, think, and swallow. The disease proceeds about 10-20 years to death. The overall prevalence of HD is 2.7/105. The incidence is higher in Europe, North America and Australia than in Asia. Prevalence in Japan and Taiwan is only 1/10 of that in Europe and North America, and there are about 30,000 to 40,000 cases in China.
Pathological Changes
In HD patients, atrophy was found in the corpus striatum, cerebral cortex, substantia nigra, thalamus, and subthalamic nucleus, with mild effects on the dentate nucleus.
The basal ganglia (or basal nuclei) are a group of subcortical nuclei, of varied origin, in the brains of vertebrates. They are associated with a variety of functions, including control of voluntary motor movements and procedural learning. The main components of the basal ganglia – as defined functionally – are the striatum, which is responsible for regulating muscle tone and coordinating various complex movements with motor and action planning. GABAergic medium spiny neurons (MSNs) in the striatum receive projections from both glutaminergic neurons in the cerebral cortex and dopaminergic neurons in the substantia nigra pars compacta.
Medium spiny neurons (MSNs) have two primary phenotypes: D1-type MSNs of the direct pathway and D2-type MSNs of the indirect pathway. The former promotes voluntary movements, while the latter inhibits involuntary movements. In the early stage of HD, a large number of MSNs are apoptotic, and the death rate of D2-type cells is greater than that of D1 type, resulting in a decrease in D2-type MSNs, which means the loss of inhibition of involuntary movements, leading to chorea. In the later stage of HD, both D1 and D2 types are impaired with the progress of the conditions. At this stage, symptoms of chorea decrease but voluntary movement is inhibited, leading to myotonia and dystonia.
Research & Therapy
Currently, there is no definitive cure for HD on the market, but some drugs may help reduce symptoms. Both Tetrabenazine and Deutetrabenazine are the inhibitors of vesicular monoamine transporter 2 (VMAT 2) in the central nervous system. Their mechanism of action (MOA) is the same: to block and inhibit the storage of monoamine neurotransmitters by targeted inhibition of VMAT 2.
Tetrabenazine
Tetrabenazine has the characteristics of short half-life and great effects, but presents some tolerability concerns in regards to adverse effects (AE). In contrast, Deutetrabenazine is a deuterated form of Tetrabenazine that exhibits significantly prolonged half-life, allowing for less frequent dosing with less side effects and good safety.
Deutetrabenazine
Valbenazine is the first drug approved by the United States Food and Drug Administration (FDA) in 2017 for the treatment of tardive dyskinesia in adults, and has a good inhibitory effect on chorea caused by HD. Pridopidine, a selective dopamine D2 receptor (D2R) antagonist, also has inhibitory effects on chorea. At present, the drug has undergone a number of clinical trials and achieved promising results. It is a new HD drug that has attracted much attention.
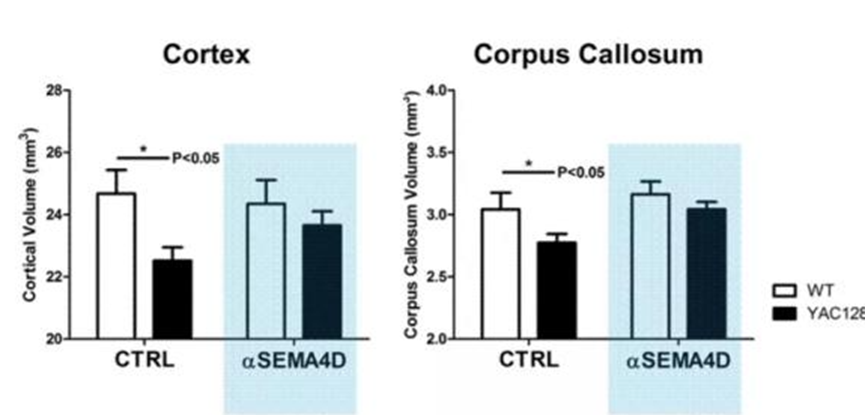
VX15, a product of Vaccinex, a biotechnology company in the United States, is a novel monoclonal antibody discovered using the ActivMAb technology platform, which can effectively block the biological activity of Semaphorin 4D (SEMA4D). SEMA4D simultaneously triggers the activation of microglial cells and astrocytes in the central nervous system. It has important regulatory functions in the pathophysiological process of various neuritis and neurodegenerative diseases such as HD and multiple sclerosis (MS), and is one of the specific molecular targets for the treatment of such diseases. Preclinical models demonstrate that the inhibition of SEMA4D can effectively alleviate neuroinflammation in the brain, thereby reducing neuronal apoptosis. In HD model mice, VX15 significantly inhibits the atrophy of cerebral cortex and corpus callosum. VX15 has been approved as an orphan drug by the FDA in 2016.
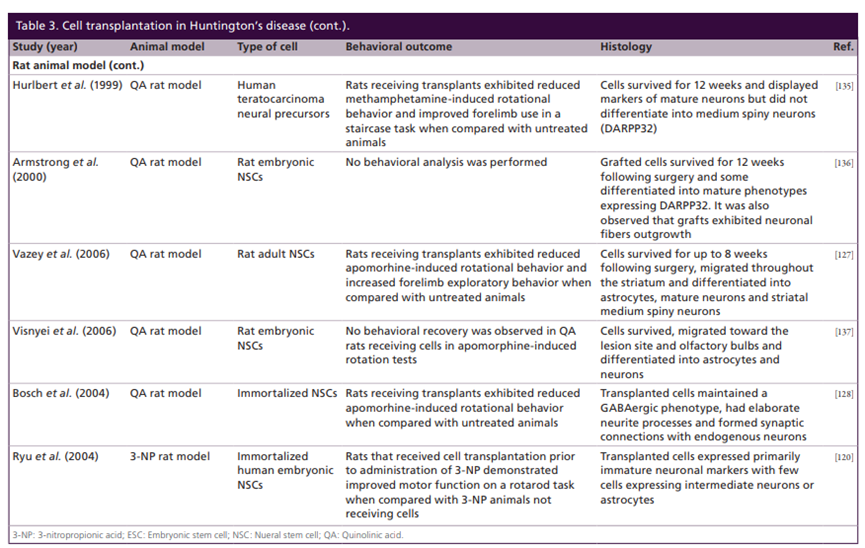
In recent years, scientists have found that stem cell therapy has the potential to repair damaged nerve tissue and replace damaged neurons to improve the condition. At present, some progress has been made in human embryonic stem cells (hESCs), human induced pluripotent stem cells (hiPSCs), and human induced neural stem cells (hiNSCs). However, this type of stem cell treatment also has certain limitations. After the stem cells are transplanted into the body, the survival and directional differentiation of the cells cannot be guaranteed. Therefore, it needs to be further explored.
In addition to these, treatments for Huntington's disease include gene therapy and another innovative treatment approach. We will introduce this progressive treatment to you in the next article, please stay tuned!
[1] Genetic Modifiers of Huntington's Disease GeM-HD Consortium. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington's Disease Onset [J]. Cell, 2019, 178(4):887-900.e14.
[2] Weijie Chen, Min Ye. Advances in Clinical Research on Early Diagnosis and Treatment of Huntington's Disease [J]. Journal of Chongqing Medical University, 2019(4):3.
[3] Ross C A , Aylward E H , Wild E J , et al. Huntington disease: natural history, biomarkers and prospects for therapeutics.[J]. Nature Reviews Neurology, 2014, 10(4).
[4] Roos R A. Huntington's disease: a clinical review[J]. Orphanet Journal of Rare Diseases, 2010, 5(1):40.
[5] Zuccato C, Valenza M, Cattaneo E .Molecular Mechanisms and Potential Therapeutical Targets in Huntington's Disease[J]. Physiological Reviews, 2010, 90(3):905-981.
[6] Deng Y P , Albin R L, Penney J B , et al..Differential loss of striatal projection systems in Huntington's disease: a quantitative immunohistochemical study.[J]. Journal of Chemical Neuroanatomy, 2004, 27(3):143-164.
[7] Caron NS, Dorsey E R, Hayden M R. Therapeutic approaches to Huntington disease: from the bench to the clinic [J]. Nature Reviews Drug Discovery, 2018, 17(10):729-750.
[8] Fink K D , Deng P , Torrest A , et al. Developing stem cell therapies for juvenile and adult-onset Huntington's disease[J]. Regenerative Medicine, 2015, 10(5):623-646.
[9] https://en.wikipedia.org/wiki/Huntington's_disease
We will respond to you in 1-2 business days.