
Retinitis pigmentosa (RP) is actually a group of inheritable, chronic eye diseases in humans that often culminate in blindness, with a clinical incidence of 1/4000. Its present-day treatment efficacy and prognosis are bleak, as it can eventually lead to blindness, which brings heavy psychological stress to patients and their families. However, ophthalmologists and researchers around the world are paying close attention to this disease and developing preventatives and treatments for patients with RP.
Pathological Study
Retinitis pigmentosa (RP) is mainly characterized by progressive degeneration of photoreceptor cells. Photoreceptor cells include rod bipolar cells and cone bipolar cells. As shown in Figure 1, rod cells are first involved in degeneration, and then secondary cone cell degeneration occurs due to biological energy supply disorders and oxidative stress, and finally histological findings are rod and cone loss.
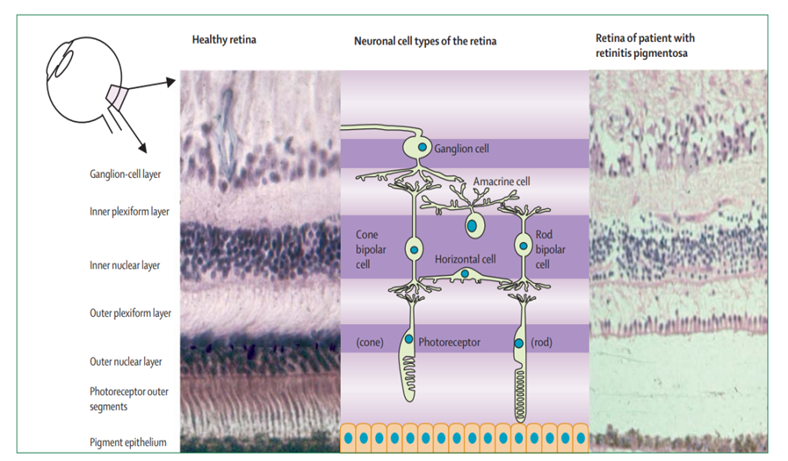
Figure 1. Histological appearance of retinas in healthy individuals (left) and retinas in patients with middle-stage RP (right).
The initial symptom of RP is decreased night vision, followed by progressive loss of peripheral visual field and development of tunnel vision (as shown in Figure 2).

Figure 2. Tunnel vision (due to loss of peripheral vision).
Eventually visual acuity gradually declines to near-blindness or complete blindness (as shown in Figure 3).
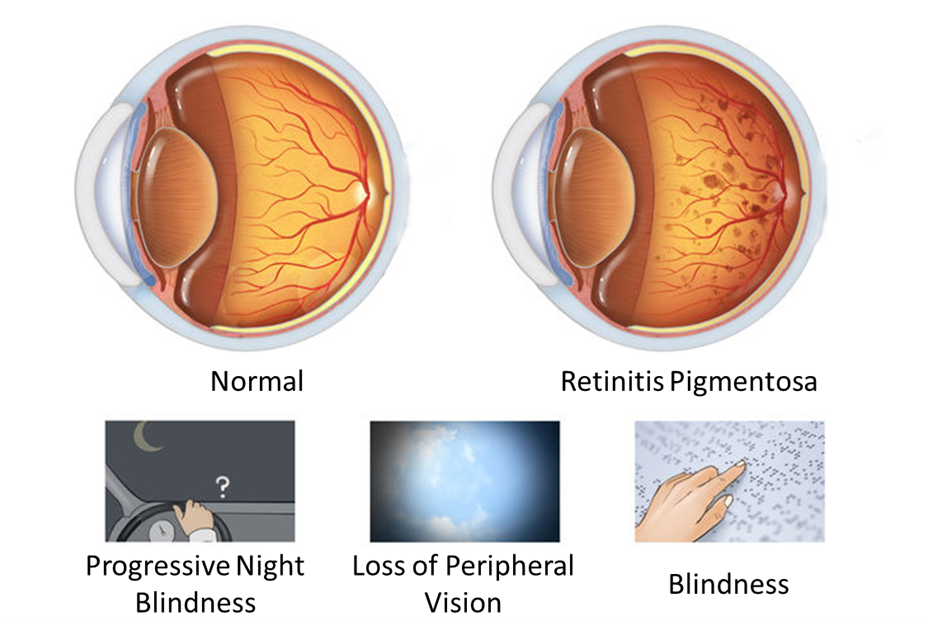
Figure 3. Changes in clinical symptoms of RP with the progression of the disease.
Findings related to RP have often been characterized in the fundus of the eye as the "ophthalmic triad" (as shown in Figure 4). This includes the development of (1) a mottled appearance of the retina and retinal pigment epithelium (RPE) that gives the same visual appearance of Bone Spicule patterns, (2) a waxy yellow appearance of the optic disk, and (3) the attenuation of blood vessels in size and Arterial/Venous ratio as they enter and exit the Optic Disk of the retina and transverse it.
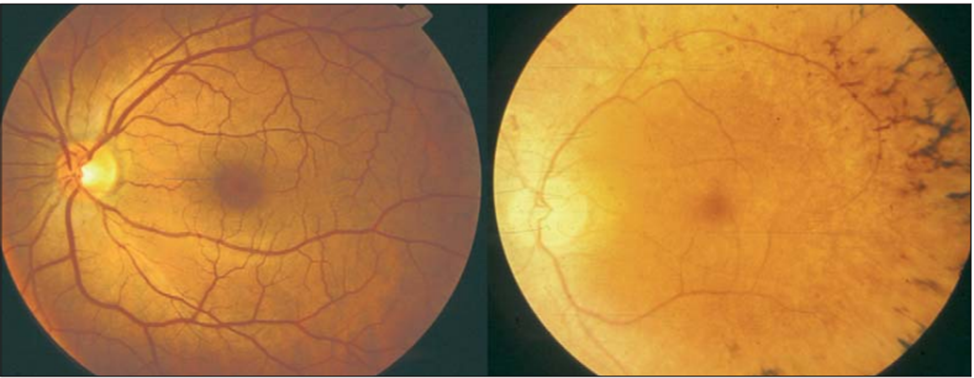
Figure 4. Fundus imaging of healthy individuals (left) and RP patients (right).
The main clinical manifestations of RP are night blindness, progressive visual field defect, and decreased visual acuity, degenerative changes in the fundus and color blindness. Some patients also experience cataracts, glaucoma, and other symptoms.
Disease-Causing Gene Mutations
In 1990, the first causative gene mutation for RP was discovered. To date, mutations in at least 79 genes have been reported to cause the disease (as shown in Table 1).
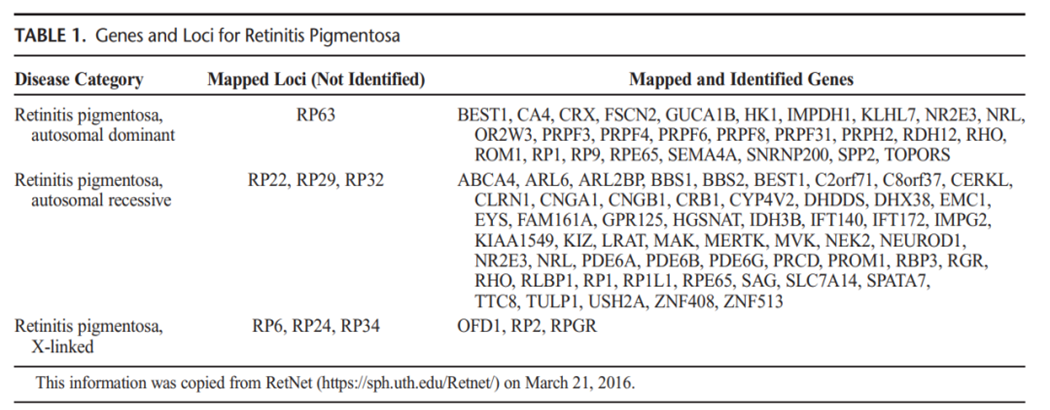
Table 1. Genes and loci for RP [4].
Different populations may have slightly different proportions of mutations in individual genes, but the major genes that cause RP are similar. Among the RP cases (caused by gene mutation) in China, 2/3 of the cases were caused by mutations in 7 genes CYP4V2, RHO, USH2A, RPGR, CRB1, RP2 and CHM.
Gene-edited animal models constructed based on information about disease-causing genes have become a valuable tool for the study of RP, not only to confirm the pathogenic role of mutations, but also to elucidate the pathophysiology of the disease. However, it is important to note that the same mutation may result in different phenotypes between mice and humans.
Animal Models of Retinitis Pigementosa (RP)
There are two categories of animal models for studying RP: one is naturally occurring animal models, and the other is artificially constructed by transgenic technology. Herein, we describe the most prevalent animal models of RP across both categories, with regard to their respective advantages and drawbacks.
Naturally occurring animal models refer to those that occur without direct manipulation or research development, and are able to replicate phenotypic traits associated with the disease pathology in humans.
No. 1 rd mice
The rd mouse is an animal model for the study of autosomal recessive retinitis pigmentosa (RP). Because of the same gene mutation and similar phenotype as the primary RP patients with autosomal recessive inheritance, this model is superior to other RP animal models and becomes an ideal research tool to explore the pathogenesis of the disease. Mice with homozygous Pde6b gene have severe RP early in life due to the nonsense mutation of the Pde6b gene, which leads to the dysfunction of the β-subunit of phosphodiesterase in the cell.
No.2 RCS rats
The RCS rat is also an animal model for the study of autosomal recessive RP. It is a relatively mature animal model of RP that was first applied to the study of the etiology and treatment of RP. RCS rats appear normal at 2 weeks after birth, and retinal photoreceptor cells developed at 17 days; the degeneration of retinal outer nuclear layer cells begin at 20 to 30 days; at 35 days, a large number of photoreceptor cells appear apoptotic; at 8 weeks, the retinal pigment epithelium (RPE) cells in the posterior pole begin to come off; at 60 days, about 99% of the photoreceptor cells degenerate; at 10 weeks, capillary endothelial cells thicken and the endothelial window disappears in the RPE-depleted area, and this multicentricity of degenerative changes occurs in different parts of the retina; about 3 months after birth, all photoreceptor cells disappear. Retinal pigment epithelial cells adjacent to degenerative photoreceptors were more numerous than normal RPE cells of the same age, with significantly more folds on the basal surface and increased mitochondria in the cytoplasm.
No. 3 rds mice
The rds mouse is an autosomal dominant RP animal model and is a classic animal model for the study of RP. Notably, rds mice cannot produce normal protein due to the insertion of a 9.2 kb genomic repeat element in the exon of the peripherin/rds gene. The expression product of Peripherin/rds gene is a structural protein of the outer segment disc membrane of vertebrate photoreceptors, which has important functions to maintain the stability and normal shape of the disc membrane.
The homozygous mutation of the Peripherin/rds gene can disrupt the production of normal disc membrane proteins, thereby further impeding the formation of the disc membrane of the outer segment of photoreceptors, which can lead to RP and hereditary macular degeneration and other retinopathy in humans. The outer segment of photoreceptor cells does not develop in homozygous mice, the outer plexiform layer and outer nuclear layer gradually become thinner from 2 weeks after birth, and retinal photoreceptor cells disappear at about 12 months after birth. Despite these phenotypes, the inner layer of the retina is largely unaffected.
No. 4 Cat and canine models
Studies have shown that cat eyes are physiologically and anatomically similar to human eyes, and the sensitivity of cats to drug doses is close to that of primates, which facilitates the experimental research of primates. The Abyssinian cat is an animal model of autosomal recessive RP, and some scholars claim that the animal model has a dominant inheritance pattern. The Abyssinian cat model shows that rods are affected in the early stage of RP, and both rods and cones are damaged in the late stage, but its genotype has not yet been confirmed. As for the canine experimental animal models, except for a very small number of X-linked inheritance models, most of them are autosomal recessive. To date, researchers have discovered many canine RP models and established some strains for experimental study.
Genetically engineered transgenic animal models refer to those that occur due to direct human intervention, and are developed to be able to replicate phenotypic traits associated with RP disease pathology in humans.
NO.1 RHO knockout mice
RHO is considered to be a gene associated with the pathogenesis of many cases of RP worldwide. There are many studies and reports on the RHO gene mutation in the world, and the reports on the pathogenesis of autosomal dominant RP are even more extensive. More than 100 RHO gene mutations have been found, of which missense mutations account for the majority.
NO.2 RPE-65 knockout mice
The retinal pigment epithelium-specific protein encoded by RPE-65 has a theoretical molecular weight of ~61 kDa (yet appears near 65 kDa on SDS-PAGE) and is widely expressed on the surface of retinal pigment epithelium. This protein is related to the metabolism of vitamin A in the retinal pigment epithelium, and is mainly involved in the metabolism of vitamin A and the regeneration of RHO. Knockout of RPE-65 gene induces retinal pigment epithelial cell dysfunction in mice, resulting in excessive accumulation of all-trans retinol and lipid deficiency of 11-cis-retinol. RPE-65 plays an important role in the reisomerization of 11-cis-retinol in the visual cycle. The inheritance pattern of RPE-65 knockout mice is mainly autosomal recessive.
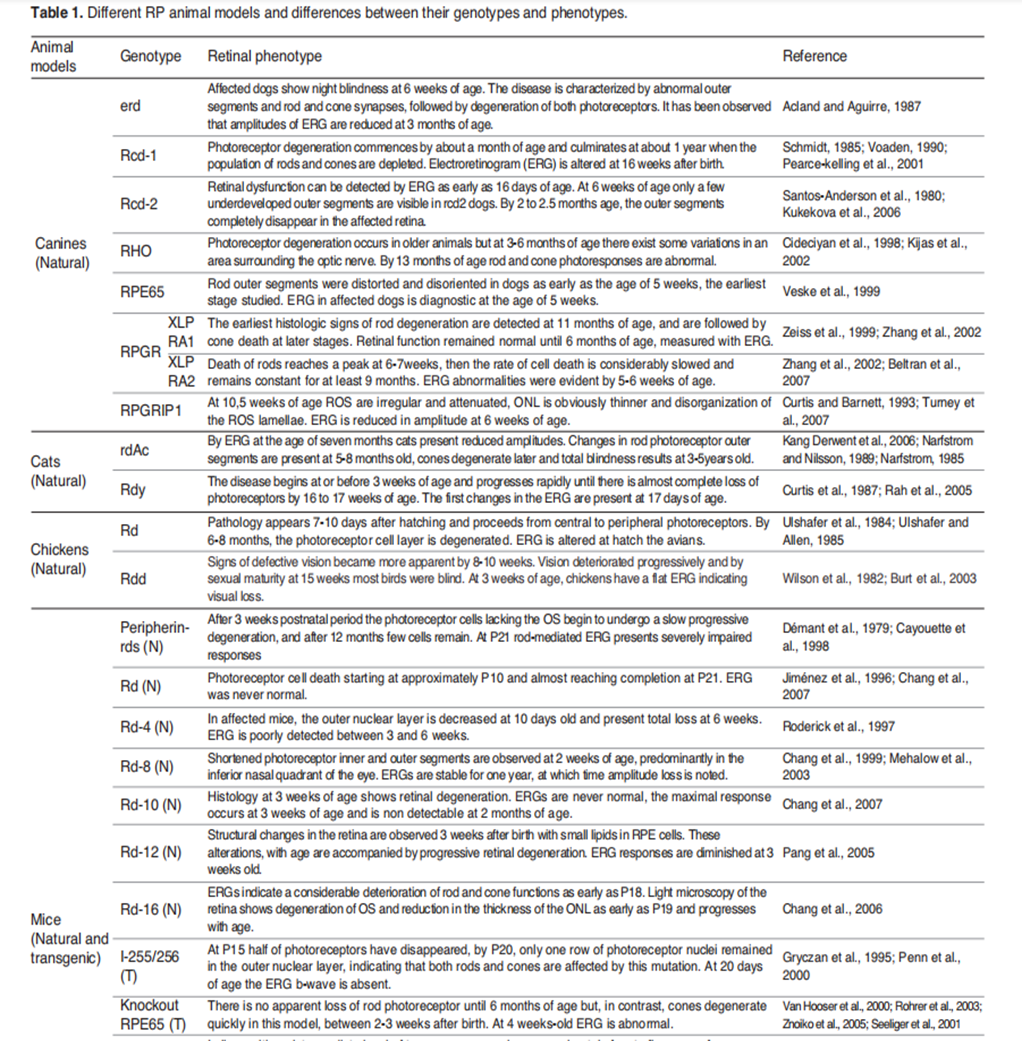
Table 2. Partial list of RP animal models. [5]
Power Your Disease Research with the RDDC

The Rare Diseases Data Center (RDDC) was developed by the Research Institute of Tsinghua, Pearl River Delta (RITPRD) with assistance from Cyagen.
The RDDC has collected and organized comprehensive data about genes, mutations, phenotypes and animal models of various types of rare diseases from international public biological resources, aiming to present them in a visual way. At present, it has collected information on over 20,000 genes and 14,000 diseases!
Users can not only better understand the information of corresponding rare diseases, but also can take advantage of various AI tools to predict pathogenicity and RNA splicing patterns, so as to help speed up the diagnosis and development of rare disease research to achieve efficient and effective therapeutics.
Preclinical Ophthalmology Research Solutions
As a comprehensive contract research organization (CRO) solution provider, Cyagen recognizes ophthalmic diseases as a breakthrough point for gene therapy and has established an ophthalmic gene therapy platform to overcome the obstacles faced by preclinical and biopharma researchers. We have equipped the platform with state-of-the-art ophthalmic instruments for small animals and an experienced professional team.
With 16 years of gene editing model construction experience, Cyagen is ready to provide you with an array of standardized and custom preclinical research solutions for ophthalmic gene therapy. If you have questions or need help with your ophthalmology drug development program, you’re invited to have a complimentary talk with our research model development experts!
[1] Zhang, Q. . "Retinitis Pigmentosa: Progress and Perspective." Asia Pac J Ophthalmol 5(2016).
[2] Wen Simin, Shao Yi, Zhou Qiong. "Progress in the pathogenesis and treatment of retinitis pigmentosa." New Advances in Ophthalmology 40.3(2020):6.
[3] A, Dyonne T Hartong , P. E. L. B. B , and P. T. P. D. A . "Retinitis pigmentosa." Lancet 368.9549(2006):1795-1809.
[4]Zhang, Q. . "Retinitis Pigmentosa: Progress and Perspective." Asia Pac J Ophthalmol 5(2016).
[5]Agurtzane Rivas, Miren, and Elena Vecino. "Animal models and different therapies for treatment of retinitis pigmentosa." Histology and histopathology (2009).
[6] Yuan Fuxiang et al. "Research progress of retinitis pigmentosa." Journal of Qingdao University: Medical Edition 55.6(2019):5.
We will respond to you in 1-2 business days.