
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common life-threatening inherited diseases. In ADPKD, fluid-filled cysts develop and persistently grow in both kidneys, eventually leading to kidney failure. Unlike some common inherited disorders, ADPKD often affects many people in a family, as it is almost always inherited by children of parents with a faulty gene. Mutations in one of the PKD1 or PKD2 genes are responsible for the majority of ADPKD cases, with mutations in PKD1 being the most common, accounting for approximately 85% of patients affected by ADPKD.
In the following, we will introduce ADPKD, its primary causative gene variant involving PKD1, and the Pkd1 knockout mice used for research.
Autosomal dominant polycystic kidney disease (ADPKD) is a common inherited kidney disease with a prevalence of 1/400-1/1000, and is the fourth leading cause of the end-stage renal disease (ESRD). The main clinical characteristic of this disease is the progressive enlargement of renal cysts accompanied by clinical manifestations such as hypertension, hematuria, urinary tract infection, kidney stones, or calcification.
ADPKD is an autosomal dominant monogenic disorder caused primarily by mutations in the PKD1 and/or PKD2 genes. Patients with PKD1 or PKD2 heterozygous mutations have a 50% probability of developing the disease in their offspring, as shown in Figure 1.
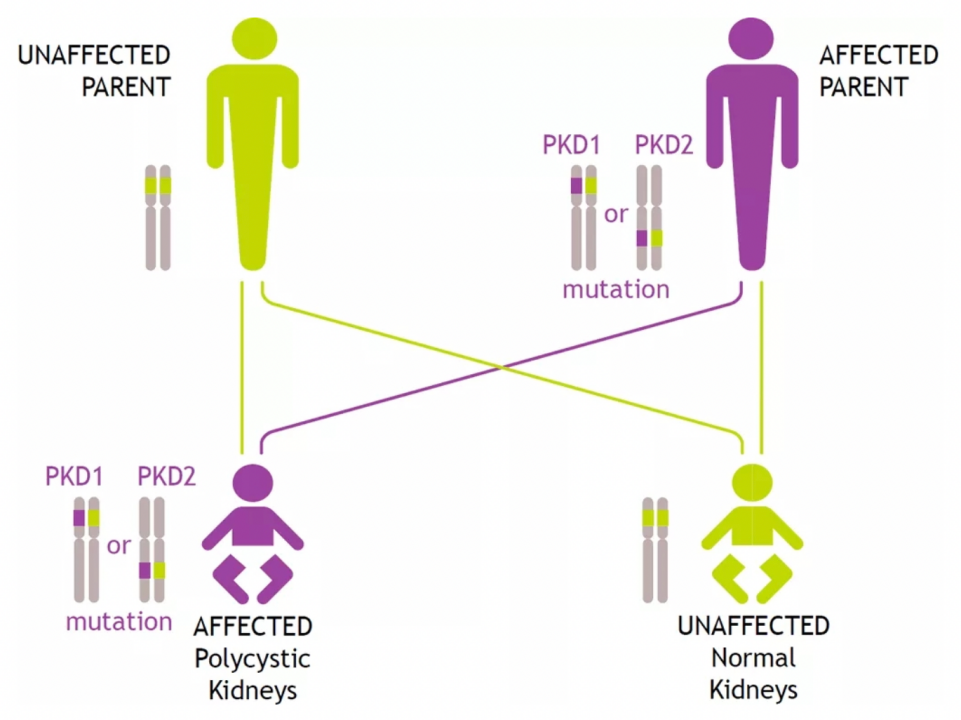
Figure 1. Genetic Family Pedigree of ADPKD[1]
Mutations in PKD1 and PKD2 account for 85% and 15% of ADPKD cases, respectively. Notably,ADPKD patients with PKD1 mutation typically have more severe disease pathology than those with PKD2 mutation. So far, 1177 PKD1 mutations and 211 PKD2 mutations have been recorded in the Human Gene Mutation Database (HGMD); 2323 PKD1 mutations and 278 PKD2 mutations have been recorded in the Autosomal Dominant Polycystic Kidney Disease Database (PKDB). Figure 2 shows the distribution of definitely pathogenic and likely pathogenic variants found in the PKD1 gene. Pathogenic mutations in PKD1 vary in type, including missense mutations, nonsense mutations, insertions and deletions, splicing mutations, and large-scale genomic rearrangements. These mutations are located in different positions of the gene and there is no discernable mutation hotspot.
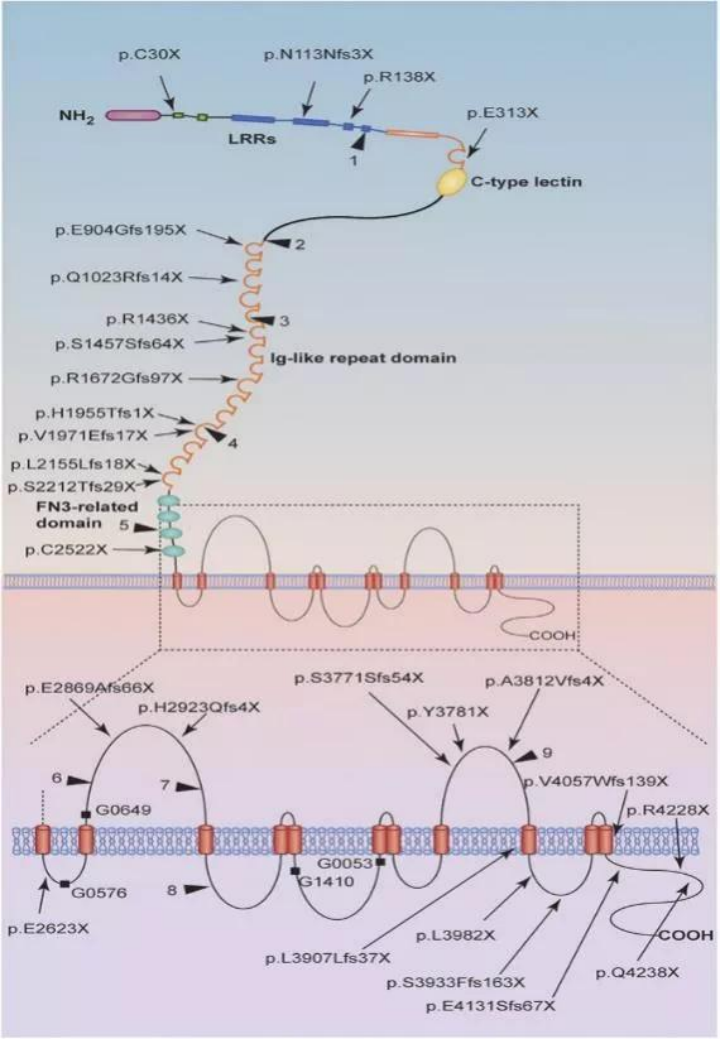
Figure 1. Schematic representation of mutations in PKD1[1]
Nonsense mutations and frameshift mutations are indicated with arrows. The insertions, deletions, and splicing mutations are indicated with boxes (G0576: c.8017-2-1delAG; G0649: c.8157-8159delCAC; G1410: c.10220 + 2T > C; G0053: c.10617 + 1delG). Likely pathogenic mutations are indicated with triangles.
The PKD1 gene (Polycystin 1, Transient Receptor Potential Channel Interacting) is located on human chromosome 16p13, with a total length of about 47kb and a total of 46 exons, encoding a transmembrane glycoprotein composed of 4302 amino acid residues - polycystin 1 (PC1).
The PC1 protein encoded by PKD1 can act as a receptor to sense external stimuli and regulate cytosolic cyclic adenosine monophosphate (cAMP) concentration and downstream signal transduction, mediating cell-cell and cell-matrix interactions. PKD1 plays an important role in renal tubular development, with mutations in this gene being closely associated with ADPKD.
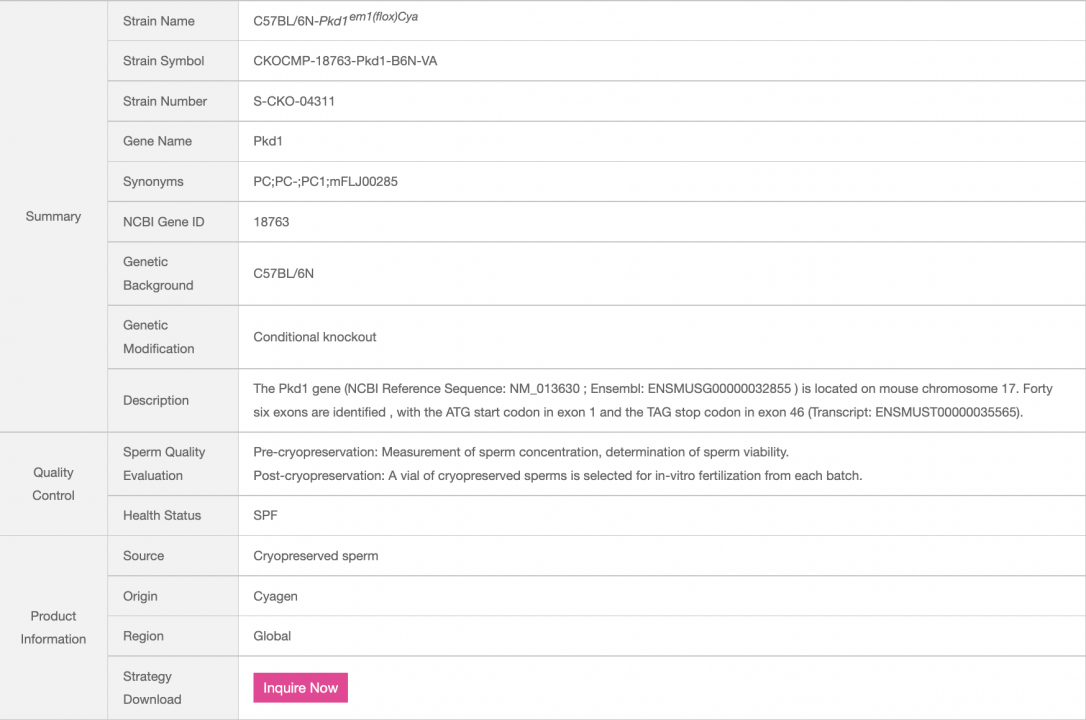
Table 1. Information on Cyagen's Pkd1 Conditional Knockout Mice
The Pkd1 homozygous knockout mice usually develop severe cysts in the kidneys and pancreas and die in infancy. In contrast, heterozygous knockout mice develop only a few cysts in the liver at later stages of development.[2]
Pionte et al. showed that knockout of Pkd1 in mice during the first 13 days of life resulted in severe renal cysts within 3 weeks. When the gene knockout was conducted in the late postnatal period, the mice did not develop cysts until 5 months later. The obtained results suggest that the pathological effects of Pkd1 inactivation depend on the state of organ development.[3]
Cabrita et al. demonstrated that deficiency of Pkd1 increases TMEM16A and cystic fiber transmembrane transducer regulator (CFTR) expression and chloride secretion in mouse kidneys, and that transepithelial chloride secreted by TMEM16A and CFTR leads to cyst enlargement. Inhibition of TMEM16A in vivo by the specific inhibitor Ani9 can largely reduce cyst enlargement and abnormal cyst cell proliferation. The results of this study provide new ideas for the treatment of ADPKD and also confirm that TMEM16A is an important therapeutic target for inhibiting the growth of cysts in ADPKD.[4]
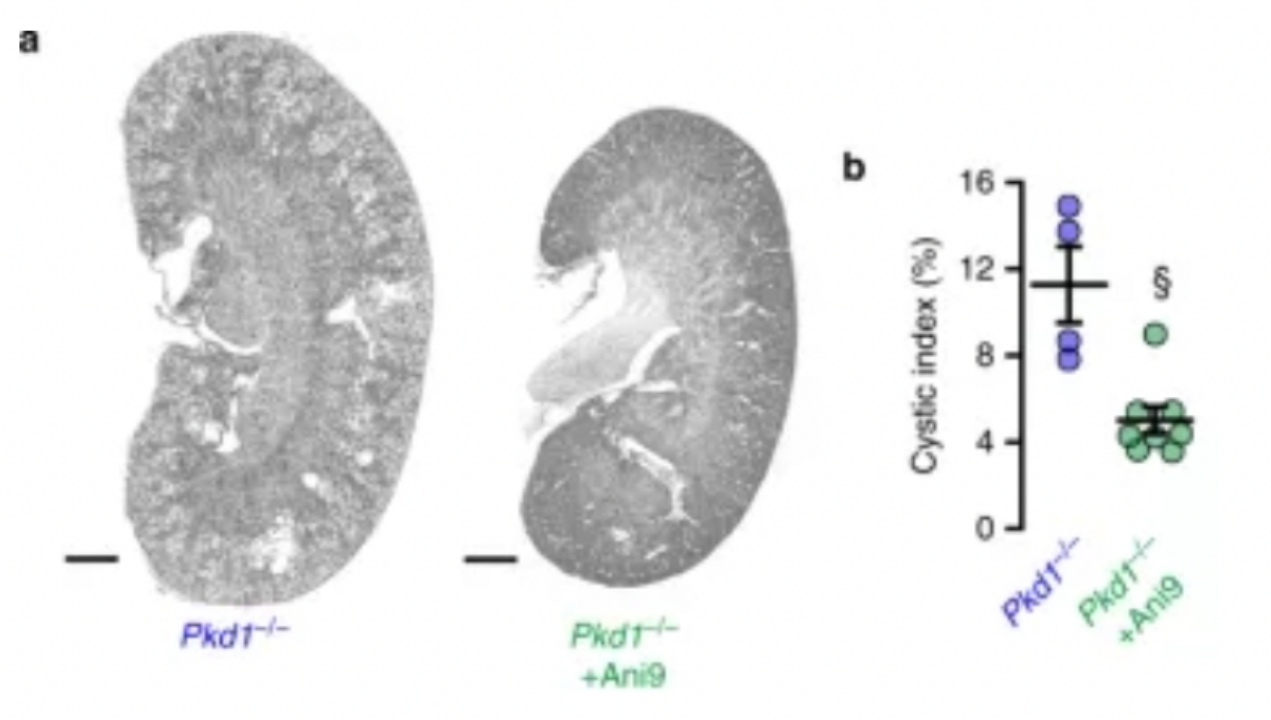
Figure 2. TMEM16A-Specific Inhibitor Ani9 attenuates renal cyst growth in vivo[4]
The diagnosis of ADPKD mainly relies on imaging examinations such as B-ultrasound, CT, and MRI. Important factors for diagnosis include family history, age of patients, and the number of bilateral renal cysts. When imaging results are inconclusive, genetic testing can be used to assist in the diagnosis.
The current clinical treatment of ADPKD focuses on relieving pain, hematuria, and symptoms caused by complications such as hypertension. If the disease progresses to end-stage renal disease (ESRD), renal replacement therapy (RRT) such as dialysis, non-dialytic care, or kidney transplantation is required. The main treatment drugs are vasopressin V2 receptor antagonist tolvaptan, the mammalian target of rapamycin (mTOR) inhibitors sirolimus and everolimus, somatostatin (SST) octreotide, and statins.
PKD1 is one of the two major causative genes of ADPKD, with PKD1 gene mutations accounting for up to 85% of all ADPKD cases. There are already more than 2,000 reported mutations of PKD1 that cause ADPKD. These disease-causing mutations are of different types, distributed in different positions of the gene, and there are no mutation hotspots, which greatly increases the difficulty of genetic analysis of ADPKD patients. Currently, no cure for ADPKD has been developed, but the symptoms of ADPKD complications can be relieved with medication. Studies have shown that inhibition of TMEM16A can greatly attenuate cyst enlargement and proliferation in mice, which provides an important therapeutic target for ADPKD. However, the efficacy of treating ADPKD with TMEM16A as a therapeutic target still needs to be tested in preclinical and clinical trials.
If you are interested in this research topic, take a look at our Knockout Catalog Models! The Pkd1 gene knockout and conditional knockout mouse strain described in this article for studying ADPKD are within our 3000+ best-selling genes, and it is deliverable in as few as two (2) weeks! Our price-matching guarantee makes sure that you can easily get the most cost-effective experimental mice for your research!
Looking for something else? We have many more gene-edited mouse models, alongside custom animal model development and breeding services. Please call 86 20-31601779 or email service-apac@cyagen.com for consultation and ordering.
[1] Liu B, Chen SC, Yang YM, et al. Identification of novel PKD1 and PKD2 mutations in a Chinese population with autosomal dominant polycystic kidney disease [published correction appears in Sci Rep. 2016;6:21578]. Sci Rep. 2015;5:17468. Published 2015 Dec 3. doi:10.1038/srep17468.
[2] Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60:321-337. doi:10.1146/annurev.med.60.101707.125712.
[3] Piontek K, Menezes LF, Garcia-Gonzalez MA, Huso DL, Germino GG. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13(12):1490-1495. doi:10.1038/nm1675.
[4] Cabrita, I., Kraus, A., Scholz, J.K. et al. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat Commun 11, 4320 (2020). https://doi.org/10.1038/s41467-020-18104-5.
[5] Cong Li, Yin Wu, Peng Li, Geng Zhang, Chunlei Li. Research progress of drugs for autosomal dominant polycystic kidney disease[J]. Drugs & Clinic, 2015, 30(9): 1167-1171.
We will respond to you in 1-2 business days.