
Genes play a significant role in the pathogenesis of eye diseases, some of which are highly prevalent in children and adults. Over 60% of cases of childhood blindness are caused by genetic factors, such as congenital glaucoma, ocular malformations, atrophy of the optic nerve, and retinitis pigmentosa. In adults, genetic factors can also be associated with serious eye diseases, including glaucoma and macular degeneration.
Reviewing Cyagen's experience in rodent model generation over the past 15 years, we have developed many mouse models for genetic variants commonly associated with human eye diseases. For example, Rho knockout (KO) mice were generated for the study of retinitis pigmentosa. In addition, Cyagen continues to develop humanized mouse models that are increasingly beneficial to disease research, including for the study of genetically-based eye diseases. In the following content, we will introduce some of the most relevant humanized mouse models for eye disease & ophthalmology research.
Retinitis pigmentosa (RP) is a clinically and genetically heterogeneous blinding retinal disease, which belongs to the large group of inherited retinal degenerations — for which 54 retinal genes have been found to be mutated thus far [1]. RP commences with progressive night blindness due to rod photoreceptor disease, accompanied by gradual loss of peripheral visual field, followed by complete blindness due to cone photoreceptor degeneration.
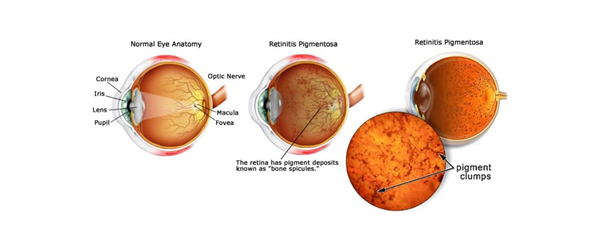
Figure 1. What is Retinitis Pigmentosa? (Source: Fleetwood Family Eye Care)
Worldwide, Retinitis pigmentosa has a prevalence of 1 in 3500, and may be inherited as an autosomal dominant (adRP), autosomal recessive (arRP), X-linked (XlRP), or digenic [2] trait. To date, there are at least 20 genes identified to cause adRP and their protein products have been implicated in diverse and critical aspects of retinal photoreceptor structure and metabolism, such as outer segment disc formation, the phototransduction and retinoid cycles, gene expression, transcription, mRNA processing, and more[3].
|
Type |
Mapped Loci (not Identified) |
Mapped and Identified Genes |
|
Autosomal Dominant Retinitis Pigmentosa (adRP) |
RP63 |
ADIPOR1, ARL3, BEST1, CA4, CRX, FSCN2, GUCA1B, HK1, IMPDH1, IMPG1, KIF3B, KLHL7, NR2E3, NRL, PRPF3, PRPF4, PRPF6, PRPF8, PRPF31, PRPH2, RDH12, RHO, ROM1, RP1, RP9, RPE65, SAG, SEMA4A, SNRNP200, SPP2, TOPORS |
|
Autosomal Recessive Retinitis Pigmentosa (arRP) |
RP22, RP29 |
ABCA4, AGBL5, AHR, ARHGEF18, ARL6, ARL2BP, BBS1, BBS2, BEST1, C8orf37, CERKL, CLCC1, CLRN1, CNGA1, CNGB1, CRB1, CWC27, CYP4V2, DHDDS, DHX38, EMC1, ENSA, EYS, FAM161A, GPR125, HGSNAT, IDH3B, IFT140, IFT172, IMPG2, KIAA1549, KIZ, LRAT, MAK, MERTK, MVK, NEK2, NEUROD1, NR2E3, NRL, PCARE, PDE6A, PDE6B, PDE6G, POMGNT1, PRCD, PROM1, PROS1, RBP3, REEP6, RGR, RHO, RLBP1, RP1, RP1L1, RPE65, SAG, SAMD11, SLC7A14, SPATA7, TRNT1, TTC8, TULP1, USH2A, ZNF408, ZNF513 |
|
X-linked Retinitis Pigmentosa (XlRP) |
RP6, RP24, RP34 |
OFD1, RP2, RPGR |
Table 1. Mapped and Identified Genes with Retinitis pigmentosa (RP) [1]
(Source: RetNet, the Retinal Information Network)
The study by Haim et al showed that the adRP form has a 8.4% prevalence across all non-atypical RP cases, arRP in 19%, X-linked in 14.3%[4] [5]. The most common single gene that causes adRP is rhodopsin (RHO; MIM# 180380), which accounts for 8% to 10% of cases with more than 130 mutations identified so far [6] [7].
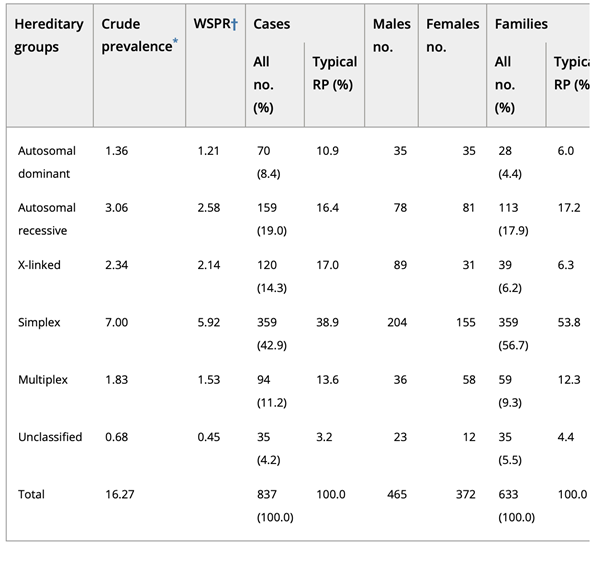
Table2. Distribution of hereditary groups in Retinitis pigmentosa (RP) [4]
RHO (Rhodopsin) is a Protein Coding gene. The rhodopsin protein encoded by the RHO gene is found in rod cells in the back of the eye and is essential for vision in low-light conditions. As part of the light-sensitive tissue at the back of the eye (the retina), rods provide vision in low light. Other light receptor cells in the retina, called cones, are responsible for vision in bright light. Rhodopsin protein (RHO) is attached (bound) to a molecule called 11-cis retinal, which is a form of vitamin A. When light hits this molecule (11-cis retinal), it activates rhodopsin and sets off a series of chemical reactions that create electrical signals. These signals are transmitted to the brain, where they are interpreted as vision.
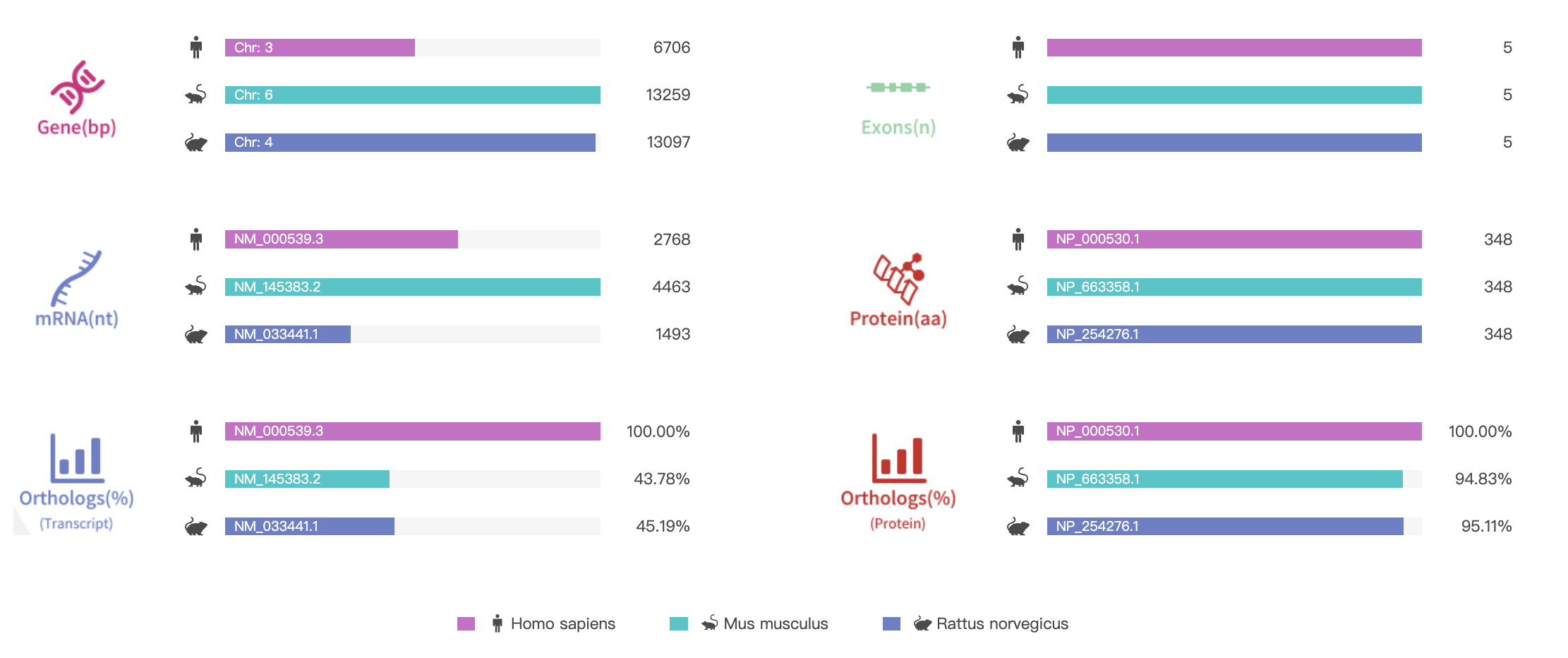
Figure 2. Gene information and sequence homology alignment of RHO [8]
Source: Rare Diseases Data Center (RDDC)
More than 150 mutations in the RHO gene have been identified in people with retinitis pigmentosa (RP). RHO gene mutations account for 20 to 30 percent of all cases of autosomal dominant RP, which is thought to be the most common form of the disorder. Rarely, mutations in the RHO gene are associated with autosomal recessive RP. However, this form of disorder usually results from mutations in other genes.
Most of the RHO gene mutations responsible for retinitis pigmentosa (RP) alter the folding or transport of the rhodopsin protein. A few mutations cause rhodopsin to be constitutively activated instead of being activated in response to light. Studies suggest that altered versions of rhodopsin interfere with essential cell functions, causing rods to self-destruct (undergo apoptosis). Because rods are essential for vision under low-light conditions, the loss of these cells leads to progressive night blindness in people with RP.
Retinitis pigmentosa is also associated with a gradual loss of cone cells, which normally provide vision in bright light. The death of cone cells leads to tunnel vision and ultimately blindness in many affected individuals. It is unclear how mutations in the RHO gene affect the function and survival of cone cells [9].
There are more than 100 RHO mutations leading to ADRP, the most prevalent RHO mutation in North America is P23H (proline-23 substituted by histidine). This was the first RP mutation to be identified, and it has been studied extensively both in vitro and in animal models. Recently, some RHO humanized mouse model strains (e.g., T17M, P23H) have been generated, which permit preclinical testing of the therapeutic effect of Targeted Gene Editing for in vivo systems without putting humans at risk. While there are few available humanized mouse models compared to the number of known RHO mutations, it is time-consuming and costly to build humanized mice for each mutation.
In one case, Xiaozhen Liu et al. used a homology-directed repair strategy combined with Targeted Gene Editing to introduce a human RHO genomic fragment containing the replacement of mouse exon1 (mE1) after the start codon to mE5 before the stop codon and replacing all introns with the human counterparts. The human rhodopsin could express itself under the control of the endogenous murine promoter both transcriptionally and translationally in vivo. Unmutated human rhodopsin in humanized mouse lines could replace murine rhodopsin morphologically and functionally. However, human rhodopsin containing T17M, G51D, G114R, R135W, and P171R mutations simultaneously in mutant humanized (Mut-Rhowt/hum and Mut-Rhohum/hum) mouse lines caused retinal degeneration [10].
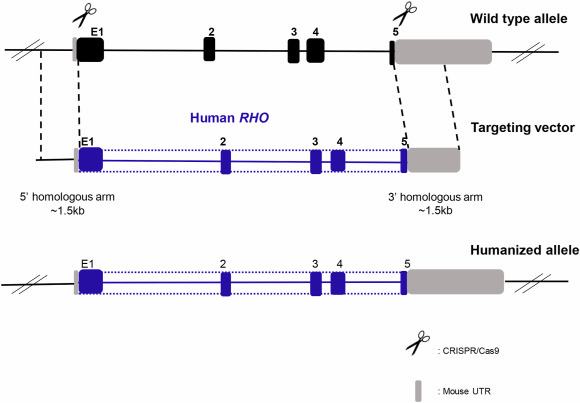
Figure 3. Humanized RHO mouse generation strategy [10]
Mut-Rhohum/hum mice suffered from severe retinal degeneration with defective formation of the rod outer segment, yielding non-recordable electroretinogram (ERG) results at 3 months. Mut-Rhowt/hum mice had a slower rate of photoreceptors loss. In 7-month-old Mut-Rhowt/hum mice, statistically reduced scotopic ERG responses were visible compared with age-matched WT mice, but the shortened outer segment and thinner outer nuclear layer could be observed from 3 months. From 7 months to 9 months, significantly abnormal scotopic ERG responses were visible and photoreceptors loss was also obvious in 9-month-old Mut-Rhowt/hum mice. In 12-month-old Mut-Rhowt/hum mice, statistically reduced scotopic and photopic ERG responses and retinal degeneration throughout the retina were visible.
The described mutant humanized heterozygotes strategy provides opportunities to explore gene editing therapies via Targeted Gene Editing for these five mutations in preclinical studies in a time-saving and cost-effective manner.
Over the past year, Cyagen has focused on building an ophthalmic gene therapy platform. We aim to provide a one-stop solution for preclinical eye disease research: from generation of ophthalmic disease models to injection of AAV, and evaluation of in vitro and in vivo pharmacodynamics.
Cyagen Knockout Catalog Models feature thousands of gene knockout (KO) mice and conditional knockout (cKO) mice. We can provide you with ready-to-use Rho knockout mice or generate customized models for you. If you need knockout (KO) and conditional knockout (cKO) mice, take a look at our best offer! Through November 30, order cKO models from Cyagen AI Knockout Mouse Model eBank and receive FREE KO – fast-track your research with complete KO/cKO cohorts for only $12,000!

>>Order now to lock-in $5,999 in savings on your KO/cKO project!
Looking for something else? We have many more gene edited rodent models and custom project capabilities. Please contact us or email service-apac@cyagen.com for consultation and ordering.
[1] RetNet. Available at: http://www.sph.uth.tmc.edu/Retnet. Accessed October 2015.
[2] Rivolta C, Sharon D, DeAngelis MM, Dryja TP. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet. 2002 May 15;11(10):1219-27. doi: 10.1093/hmg/11.10.1219. Erratum in: Hum Mol Genet. 2003 Mar 1;12(5):583-4. PMID: 12015282.
[3] Hims MM, Diager SP, Inglehearn CF. Retinitis pigmentosa: genes, proteins and prospects. Dev Ophthalmol. 2003;37:109-25. doi: 10.1159/000072042. PMID: 12876833.
[4] Haim M. x. 2002;(233):1-34. doi: 10.1046/j.1395-3907.2002.00001.x. PMID: 11921605.
[5] Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010 Apr;11(4):273-84. doi: 10.1038/nrg2717. PMID: 20212494.
[6] Briscoe AD, Gaur C, Kumar S. The spectrum of human rhodopsin disease mutations through the lens of interspecific variation. Gene. 2004 May 12;332:107-18. doi: 10.1016/j.gene.2004.02.037. PMID: 15145060.
[7] HGDB-Cardiff. Available at: http://www.hgmd.org. Accessed October 2015.
[8] RDDC. Available at: https://rddc.tsinghua-gd.org/details/gene?gene=xnEBPG.
[9] GeneCards. Available at: https://www.genecards.org
[10] Xiaozhen Liu, Ruixuan Jia, Xiang Meng, Ying Li, Liping Yang,
Retinal degeneration in humanized mice expressing mutant rhodopsin under the control of the endogenous murine promoter, Experimental Eye Research, Volume 215, 2022, 108893, ISSN 0014-4835, https://doi.org/10.1016/j.exer.2021.108893.
We will respond to you in 1-2 business days.