
Different types of mutations in genes can affect the body in different ways; Sometimes certain mutations in genes can cause the protein to not work or be made at all. Cystic fibrosis is caused by mutations in the gene encoding cystic fibrosis transmembrane conductance regulator (CFTR). This protein is responsible for regulating the secretion and absorption of ions and water in epithelial tissues. In the previous article, we introduced cystic fibrosis (CF), a genetic disease. This article will discuss the CFTR gene, its 6 types of mutations, and the application of the CFTR mouse model in research.
Cystic fibrosis (CF) is a common autosomal recessive disease caused by mutations in the CFTR gene, which mainly affects epithelial tissues, such as airway epithelial cells, small intestine, pancreas, liver, salivary glands, reproductive tract, etc.
The CFTR gene is located on the long arm of chromosome 7 (7q31.2), with a full length of 250 kb, 1480 amino acids, 27 exons and 26 introns. The CFTR gene encodes the cystic fibrosis transmembrane conductance regulator (CFTR) protein consisting of 12 transmembrane domains, 2 nucleotide-binding domains (NBD1/2), and a regulatory (R) domain. The R-domain contains phosphorylation sites shared by protein kinase A (PKA) and protein kinase C (PKC), and when protein kinases are phosphorylated, the CFTR chloride channel is opened.
CFTR proteins are members of the ATP-binding cassette (ABC) transporters family, which are involved in the uptake of nutrients, export of substrates, and mediating intercellular communication in eukaryotes and bacteria. It is expressed in the mucous glands, drives the secretion of fluids and electrolytes, and secretes mucins, glycoconjugate, and antimicrobials from the mucous glands into the respiratory tract to facilitate mucociliary clearance and self-defense.
Mutations in CFTR cause abnormal activation of chloride channels, affecting the regulation of salt ions and water uptake by cells. As a result, the mucus in the channel becomes viscous, causing embolisms in organs such as the pancreas and respiratory tract. Additionally, sodium chloride in the sweat ducts cannot be reabsorbed, resulting in very salty sweat.
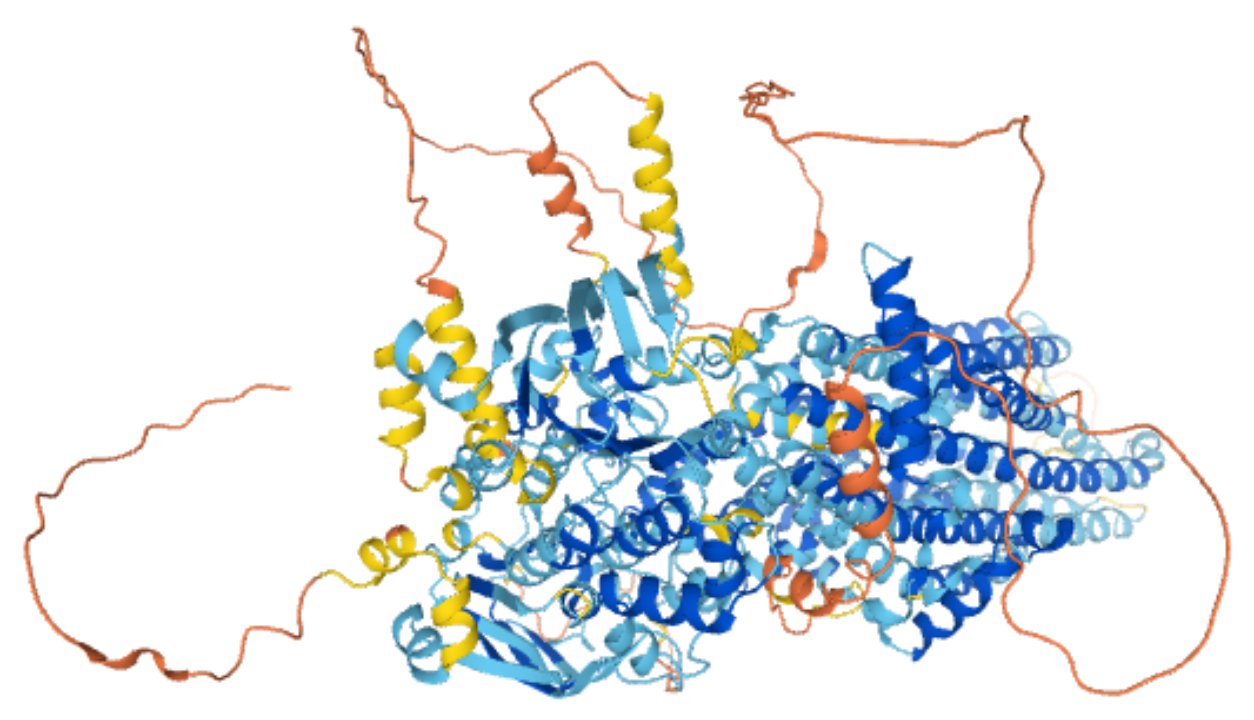
Figure 1. Prediction of CFTR Protein Structure
Activation of CFTR is mediated by cyclic adenosine monophosphate (cAMP), and its nucleotide-binding domain (NBD) binds to adenosine triphosphate (ATP), but some studies have pointed out that it can also be achieved independently of cAMP.
CFTR plays an important role in the regulation of ion channels, mediating the movement of intracellular chloride ions across the membrane, accompanied by the transport of water molecules. The process by which CFTR regulates chloride channels is as follows:
In addition to mediating the efflux of chloride ions, CFTR can:
In normal CFTR-sufficient cells, the functional CFTR protein is correctly trafficked to the plasma membrane. Over 2000 CFTR mutations have been identified to date and are divided into 6 categories.
Class I mutations result in a lack of CFTR protein synthesis.
Class II mutation is the most common type of CFTR mutation. The mutation is caused by a 3 base pair deletion in exon 10 resulting in a deletion of phenylalanine at position 508 of the protein. CF patients with △F508 mutation accounted for 88.5% of the total. The mutations result in a block in CFTR maturation and the degradation of the misfolded protein in the endoplasmic reticulum (ER).
Class III mutations affect the regulation of the CFTR, where the CFTR channel is less functional.
Class IV mutations may cause the reduction of Cl− conductance.
Class V mutations lead to reduced synthesis of functional CFTR.
Class VI mutations lead to decreased stability of CFTR at the cell membrane.
The CFTR amino acid sequence homology between human and mouse is 78%. The first CFTR knockout mouse, CFTRtm1UNC, was generated by the insertion of a stop codon in exon 10 of the CFTR coding sequence, termed the S489X mutation, which has severe intestinal complications.
There are more CFTR knockout mouse models on various genetic backgrounds including the CFTRtm1CAM, CFTRtm1HSC, CFTRtm1BAY, and CFTRtm3BAY models. None of these mouse strains had detectable mRNA for wild-type CFTR, CFTRtm1CAM had an intestinal phenotype with severe pancreatic duct obstruction, and CFTRtm1HSC, CFTRtm1BAY, and CFTRtm3BAY all had severe intestinal complications.
In addition to the knockout model of CF, various knock-in mutants were also developed, such as the generation of mouse models carrying the ΔF508 mutation (CFTRtm1EUR, CFTRtm1KTH, CFTRtm2CAM), CFTRtm2HGU carrying the G480C mutation, and CFTRtm1G551D carrying the G551D mutation. Among them, the CFTRtm1EUR model has a non-lethal intestinal abnormal phenotype, but no abnormality in the pancreas and liver; the sperm of the CFTRtm1KTH model has an abnormal phenotype, but no abnormality in the gallbladder and pancreas, and has a longer survival period than knockout mice.
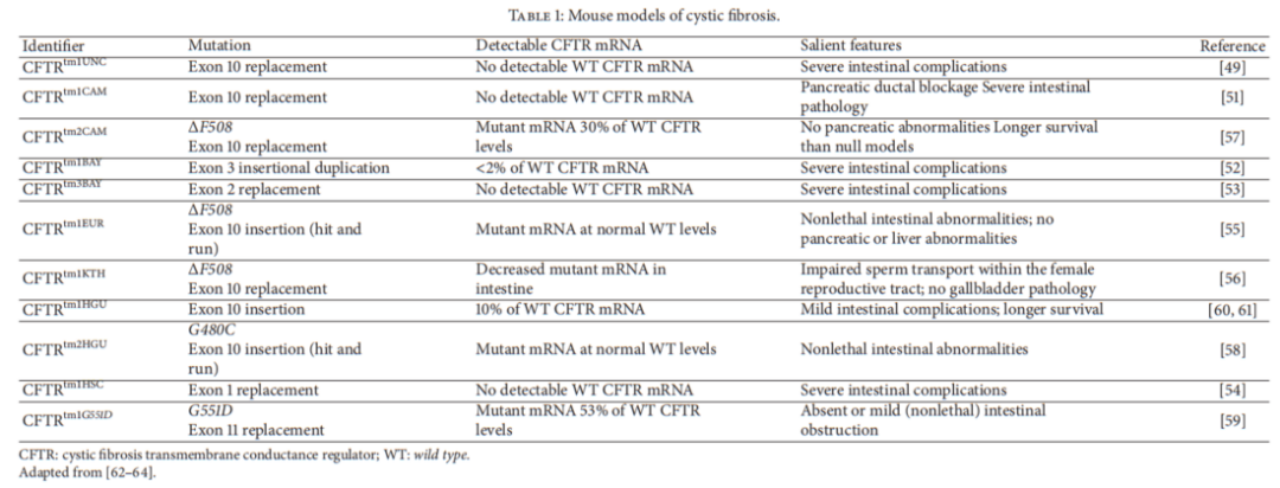
Figure 2. Mouse models of cystic fibrosis.
In the study of cystic fibrosis, knockout and knock-in mouse models have the advantages of easy construction, short breeding period, and low cost of feeding, but they still have certain limitations due to the inconsistent disease severity of models of different species. For example, the above-mentioned mouse model can show severe intestinal obstruction, growth inhibition, and olfactory electrophysiological abnormalities similar to patients, but no spontaneous pulmonary infection. Most of the experimental results showed that only minor complications occurred in the pancreas, liver, and vas deferens of mice, which were very different from the symptoms of patients.
The reason for this discrepancy may be that in mice, low-to-moderate expression of CFTR produces sufficient ion transport capacity to render the disease less severe. Also, in mice, the epithelial expression of a second Cl− channel with an alternative signaling pathway to CFTR may compensate for dysfunctional CFTR.
Therefore, some researchers tried to overcome this limitation by transferring bacterial artificial chromosomes (BAC) containing the human CFTR gene into CFTR knockout mice. The results showed that the abnormal phenotype of the intestinal and respiratory tract of mice expressing human CFTR was similar to that of patients. It also proved that the CFTR gene has a certain degree of conservation between mice and humans.
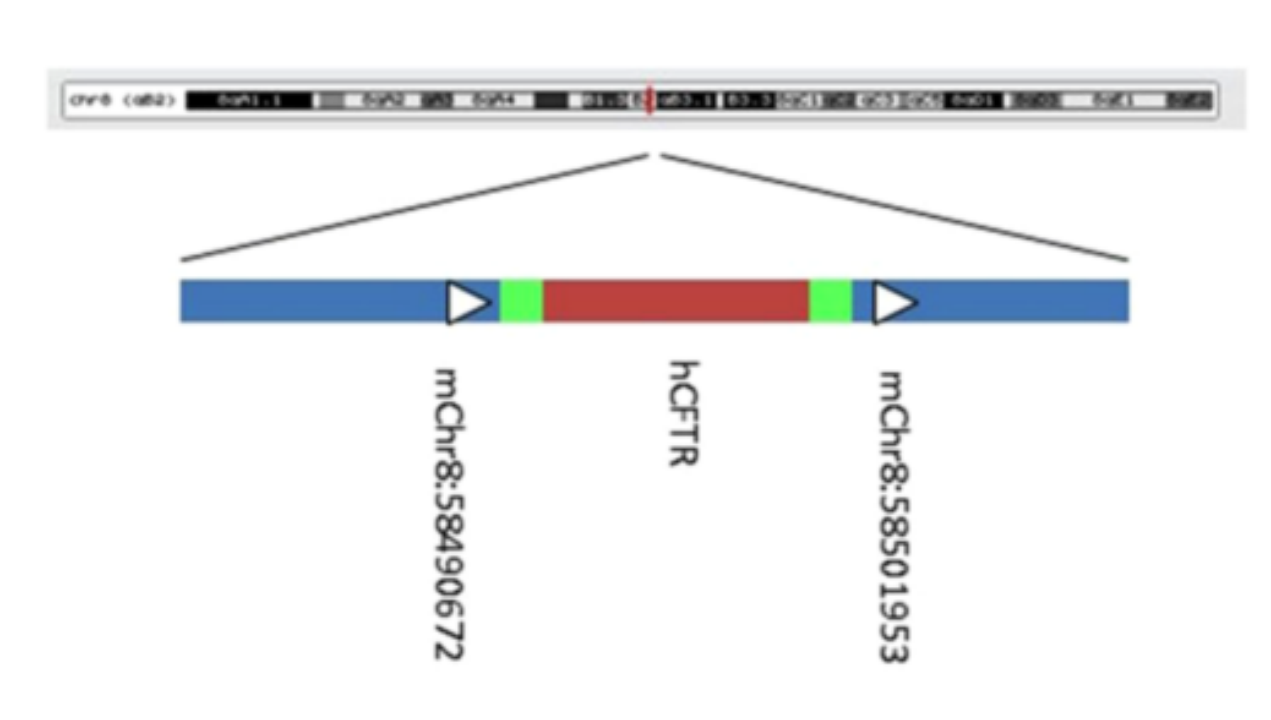
Figure 3. Construction of humanized mice.
The researchers replaced the green box region (Figure 3) in the 5'-untranslated region (5'-UTRs) of CFTR with 5'-UTRs labeled with luciferase and judged the function of the sequence by the fluorescence of luciferase. The results showed that when the upstream AUG codon (uAUG) was mutated to the UUG codon (uUUG), and the sequence of the lower stem was mutated, the luciferase expression efficiency of the mutant sequence was significantly higher than that of the 5'UTR sequence of wild-type CFTR. Also, similar results were found when mutating the sequence of the top stem. The above findings suggest the potential therapeutic value of the 5' UTR of CFTR.
Numerous miRNA binding sites have also been reported to be found on CFTR mRNA. It has been shown that miR-145-5p can inhibit the synthesis of CFTR by interacting with CFTR and other factors, and the peptide nucleic acids (PNAs) R8-PNA-a145 targeting miR-145-5p could inhibit the expression of miR-145-5p and upregulate the expression of CFTR mRNA. The above findings suggest the potential therapeutic value of the 3' UTR of CFTR.
If you are interested in this research topic, take a look at our Knockout Catalog Models! The CFTR gene knockout and conditional knockout mouse strain described in this article for studying CF are deliverable in as few as 7-9 weeks! Our price-matching guarantee makes sure that you can easily get the most cost-effective experimental mice for your research!
Looking for something else? We have many more gene-edited mouse models, alongside custom animal model development and breeding services. Please email service-apac@cyagen.com for consultation and ordering.
[1] Lavelle GM, White MM, Browne N, McElvaney NG, Reeves EP. Animal Models of Cystic Fibrosis Pathology: Phenotypic Parallels and Divergences. Biomed Res Int. 2016;2016:5258727.doi:10.1155/2016/5258727. Epub 2016 Jun 1.
[2] Rosen BH, Chanson M, Gawenis LR, Liu J, Sofoluwe A, Zoso A, Engelhardt JF. Animal and model systems for studying cystic fibrosis. J Cyst Fibros. 2018 Mar;17(2S):S28-S34. doi: 10.1016/j.jcf.2017.09.001. Epub 2017 Sep 19.
[3] Gawenis LR, Hodges CA, McHugh DR, Valerio DM, Miron A, Cotton CU, Liu J, Walker NM, Strubberg AM, Gillen AE, Mutolo MJ, Kotzamanis G, Bosch J, Harris A, Drumm ML, Clarke LL. A BAC Transgene Expressing Human CFTR under Control of Its Regulatory Elements Rescues Cftr Knockout Mice. Sci Rep. 2019 Aug 14;9(1):11828. doi: 10.1038/s41598-019-48105-4.
[4] Fabbri E, Tamanini A, Jakova T, Gasparello J, Manicardi A, Corradini R, Sabbioni G, Finotti A, Borgatti M, Lampronti I, Munari S, Dechecchi MC, Cabrini G, Gambari R. A Peptide Nucleic Acid against MicroRNA miR-145-5p Enhances the Expression of the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) in Calu-3 Cells. Molecules. 2017 Dec 29;23(1):71. doi: 10.3390/molecules23010071.513.html
We will respond to you in 1-2 business days.