
Since ancient times, children around the world have suffered from cystic fibrosis, which shortens life spans. In medieval Europe, these children were believed to be cursed by witches and doomed to death. The curse that became folklore was: "Woe to the child who tastes salty from a kiss on the brow, for he is cursed and soon will die." Salty skin was a sign of an impending disease without cause or cure. Until relatively modern times, cystic fibrosis was poorly understood beyond its folkloric diagnosis.[1] In the following, we will uncover the mystery of this inherited rare disease, which, although rare, is the most common life-limiting recessive genetic disorder among Caucasians.
Cystic fibrosis (CF) is an inherited disease that can cause severe damage to the lungs, digestive system and other organs in the body. Since cystic fibrosis transmembrane regulator protein is located in the apical plasma membrane of epithelial cells of various glands, the disease affects the cytosol produced by different types of secretory cells. As a result, the patient's secretions can become sticky and thick, unable to act as lubricants, plug up tubes, ducts and passageways, especially in the lungs or pancreas.[2]
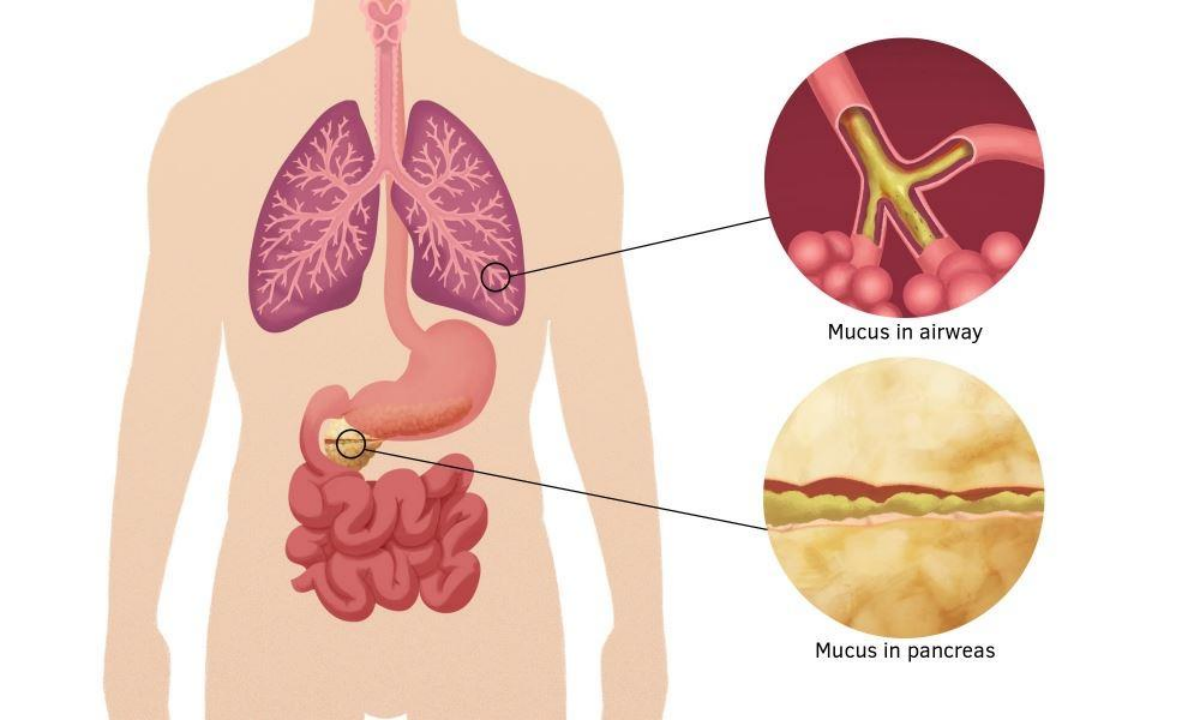
Figure 1. CF results in a build-up of thick mucus in organs such as the lungs and pancreas.[8]
CF is a progressive disease that often requires routine care and can severely impact quality of life. However, with the iterative progress of various medical technologies, the quality of life of patients has been greatly improved. Most of the patients are now able to live normally and their life expectancy has been greatly increased.
In the late 1930s, American pathologist Dorothy Andersen discovered that some children had cystic dilatation of the pancreatic duct and extensive fibrosis during the process of autopsies with malnourished children. She then described the phenomenon as "cystic fibrosis of the pancreas," from which it was discovered and named. Other scientists soon discovered that this is an autosomal recessive disorder that affects not only the pancreas but also multiple organs in the human body, including the digestive tract and lungs. Because of this, it is often confused with other diseases in clinical practice.
In 1949, a sudden heat wave brought new discoveries. Pediatric pathologist Paul di Sant’Agnese, who was caring for children with CF in the hospital at the time, noticed that some patients were experiencing severe heatstroke and dehydration. Through further research, he found it was because the percentage of salt in the patient's sweat was significantly higher than that of normal people. Based on this symptom, in 1959 Gibson et al. established a quantitative pilocarpine iontophoresis method for the diagnosis of CF.
It is simple and easy to operate, and has high sensitivity and specificity, so Gibson’s method is used as an authoritative standard for the diagnosis of CF. When the concentration of chloride ion in sweat is >60 mmol/L, the result is positive. Although this is a good diagnostic method, it is not applicable when the clinical manifestations are atypical, the concentration of chloride ions in sweat is in the middle range, or when the baby is less than 48 hours old.
Cystic fibrosis is an autosomal recessive disorder that is most common in the Caucasian population. The incidence of the disease is 1/2500-1/3500 in newborns, 1/3500 in the US population, and lower in Asia.[3] Up to one in 25 individuals of Northern European ancestry is considered a genetic carrier. The disease appears only when two of these carriers have children, as each pregnancy between them has a 25% chance of producing a child with the disease.[4]
In some countries, newborns are genetically screened to confirm whether they have CF within a month following birth, before they show typical symptoms. Unscreened newborns may not be diagnosed until signs and symptoms appear, and some patients may not develop symptoms until their teens or adults.
Signs and symptoms of CF mostly affect the respiratory and digestive systems and vary depending on the severity of the disease. Even in the same person, symptoms may worsen or improve over time. Patients diagnosed in adulthood are usually less ill and more likely to experience atypical symptoms such as recurrent pancreatitis, infectious pneumonia, and infertility.
The viscous fluid associated with CF can plug up the tubes that carry air to the lungs, which can trigger the following signs and symptoms:
Thick mucus can also plug up the digestive tract, preventing the transport of digestive enzymes from the pancreas to the small intestine, which results in the intestinal tract being unable to fully absorb nutrients from food. Eventually the following symptoms may occur:
CFTR gene mutation is mostly manifested with congenital absence of bilateral or unilateral vas deferens and decreased sperm quality in male infertility. Female patients can be fertile, but special attention to lung capacity is required during pregnancy.
Nearly 40% of patients have anemia, of which, about 10% have severe anemia. Chronic infections can also lead to anemia. Some patients have the triad of hypoalbuminemia, edema, and anemia.
The thick mucus in the lungs and sinuses is the best place for bacteria and fungi to grow. People with CF often have sinus infections, bronchitis, or infectious pneumonia, as well as intractable bacterial infections that are resistant to antibiotics.
The lining of the nose becomes inflamed and swollen, and soft granulation-like growths (polyps) develop.
Bronchiectasis lesions are found near the blood vessels in the lungs. Respiratory damage combined with infection may lead to hemoptysis. The blood volume is small, but it can still be life-threatening.
CF can severely damage lung tissue, preventing it from functioning properly. This symptom usually manifests as a progressive deterioration of lung function, which can eventually be life-threatening. Respiratory failure is the most common cause of death.
The pancreas secretes insulin, which the body needs to use sugar. CF increases the risk of developing diabetes, which occurs in about 20 percent of adolescents and 40 to 50 percent of adults.
Thick mucus can plug up the tubes that carry digestive enzymes from the pancreas to the intestines. Without these enzymes, the body cannot absorb protein, fat, or fat-soluble vitamins to get enough nutrients, which can lead to growth retardation, weight loss, or inflammation of the pancreas.
Patients are at high risk of developing dangerous osteoporosis and may also experience joint pain, arthritis, and myalgia.
Chronic diseases can cause distress and emotions such as fear, depression, and anxiety, which can further impact the patients’ quality of life and even physical condition.
CF is usually diagnosed through a variety of tests, including physical examination, checking for symptoms, and gene detection.
Women who are pregnant or couples planning a pregnancy can have themselves tested for the CFTR gene mutations to determine the risk that their child will be born with CF.[5] It can also be determined by testing for trypsinogen (IRT) released from the pancreas, but this result can fluctuate due to premature or stressful delivery.
Newborns older than 2 weeks may undergo a sweat test. An electric current is used to drive pilocarpine into the skin, stimulating sweating. The sweat is collected and analyzed for salt levels. Having unusually high levels of chloride in the sweat suggests CFTR is dysfunctional; the person is then diagnosed with cystic fibrosis.[6]
Genetic detection and sweat tests are recommended in patients with recurrent pancreatitis, nasal polyps, chronic sinus or lung infections, bronchiectasis, and in patients with infertility.
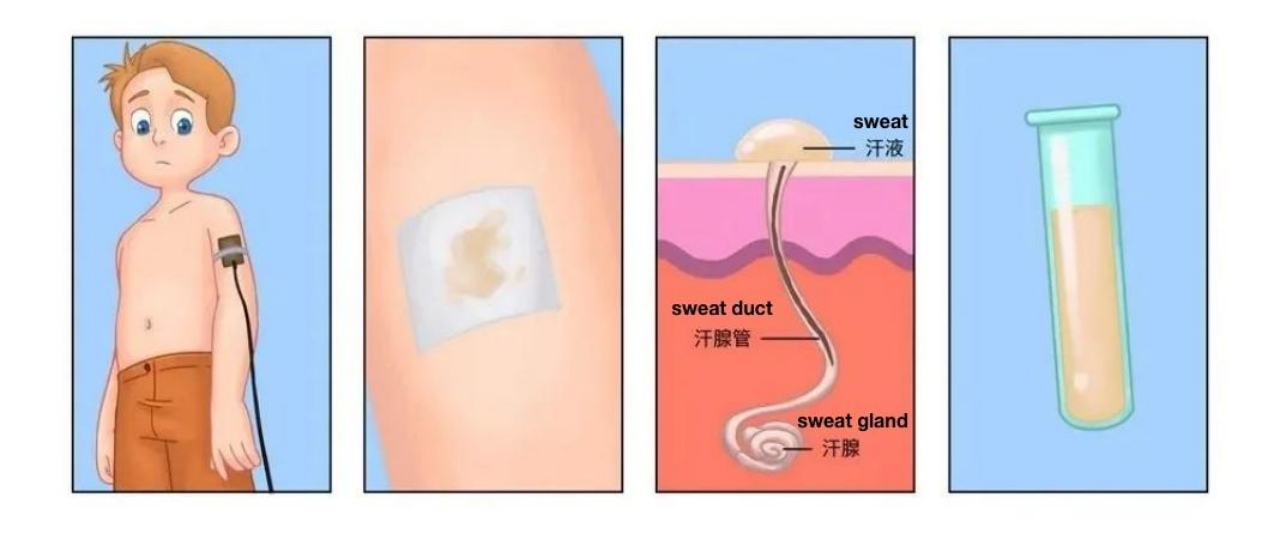
Figure 2. Sweat Test
For example, nasal surgery, oxygen therapy, non-invasive ventilation, esophagus, lung or liver transplantation.
The Rare Diseases Data Center (RDDC) was developed by the Research Institute of Tsinghua, Pearl River Delta (RITPRD) with assistance from Cyagen Biosciences.
The RDDC database has collected and sorted out all information about rare diseases from international public biological resources, including related genes, mutations, phenotypes, and animal models of rare diseases, aiming to present them with added visualization capabilities. At present, the RDDC has collected information on over 20,000 genes and 14,000 human diseases - all freely accessible!
Users are not only able to better understand the information of corresponding rare diseases, but also can take advantage of various AI tools to predict pathogenicity and RNA splicing patterns. The RDDC tools are available to help speed up the process of research and development of treatments for rare diseases. Try out the AI Tools for Genetic Analysis now:

Click the photo above for more information!
*Statement: RDDC data and tools are only for scientific research reference, and should not be used as the final conclusion in medical diagnosis and evaluation.
[1] Yu E, Sharma S. Cystic Fibrosis. 2022 Aug 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 29630258.
[2]https://www.mayoclinic.org/zh-hans/diseases-conditions/cystic-fibrosis/symptoms-causes/syc-20353700
[3] http://endo.dxy.cn/article/140307
[4] Edwards QT, Seibert D, Macri C, Covington C, Tilghman J (November 2004). "Assessing ethnicity in preconception counseling: genetics--what nurse practitioners need to know". Journal of the American Academy of Nurse Practitioners. 16 (11): 472–80. doi:10.1111/j.1745-7599.2004.tb00426.x. PMID 15617360. S2CID 7644129.
[5] "Carrier Screening in the Age of Genomic Medicine". American College of Obstetricians and Gynecologists. 2017. Archived from the original on 25 February 2017. Retrieved 22 February 2020.
[6] Egan, Schechter & Voynow 2020, "Diagnosis and Assessment".
[7] Rowe SM, Clancy JP, Wilschanski M. Nasal potential difference measurements to assess CFTR ion channel activity. Methods Mol Biol. 2011;741:69-86. doi: 10.1007/978-1-61779-117-8_6. PMID: 21594779; PMCID: PMC3760477.
[8] https://app.healthand.com/jp/topic/general-report/cystic-fibrosis
We will respond to you in 1-2 business days.